MODIFIED TEMPLATE-INDEPENDENT ENZYMES FOR POLYDEOXYNUCLEOTIDE SYNTHESIS
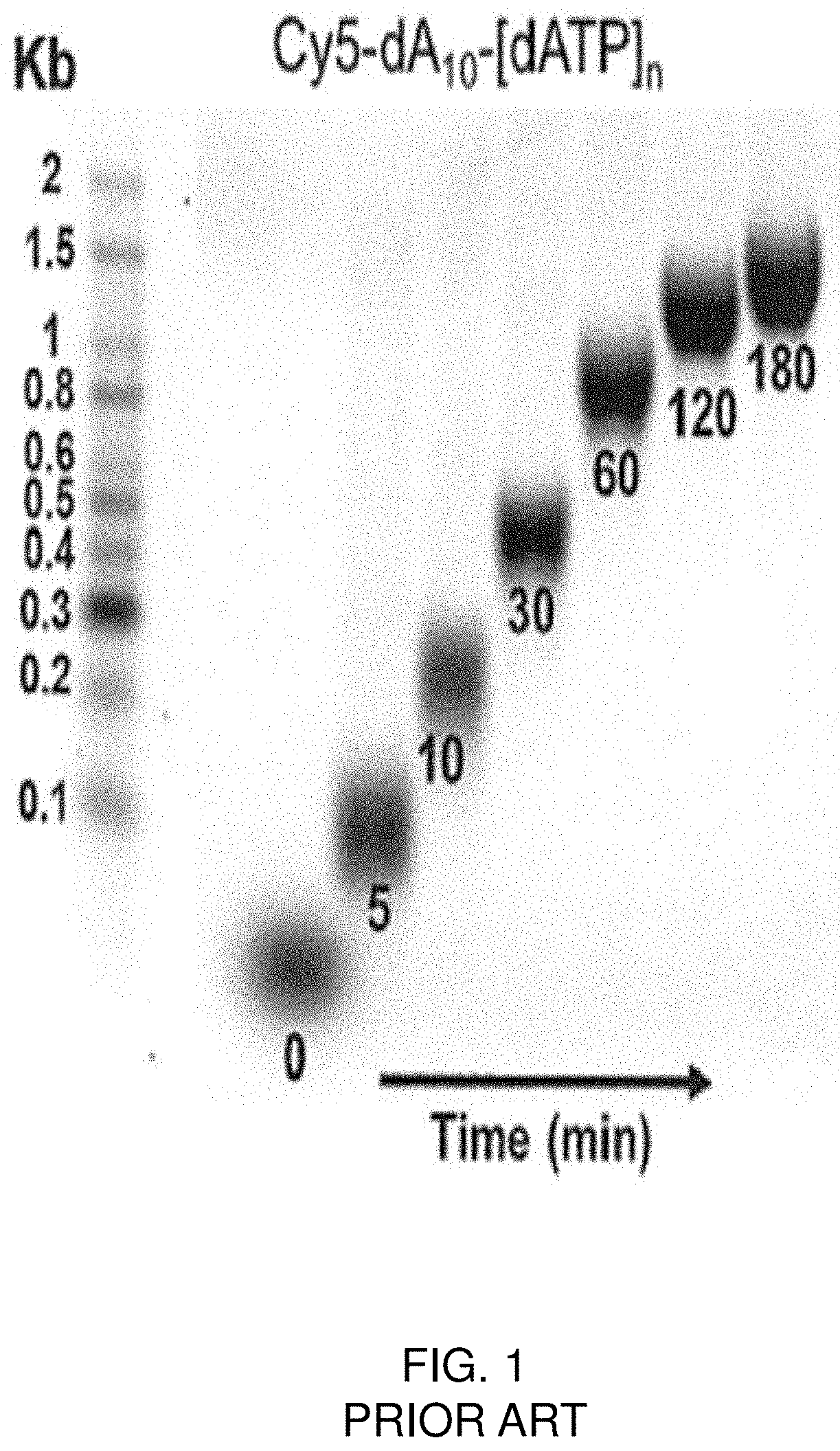
This application is a continuation-in-part of U.S. Non-Provisional application Ser. No. 16/165,465, filed Oct. 19, 2018, which is a continuation-in-part of U.S. Non-Provisional application Ser. No. 16/113,757, filed Aug. 27, 2018, which is a continuation of U.S. Non-Provisional application Ser. No. 14/918,212, filed Oct. 20, 2015, now issued as U.S. Pat. No. 10,059,929, which claims priority to and the benefit of U.S. Provisional Application Ser. No. 62/065,976, filed Oct. 20, 2014, the content of each of which is incorporated by reference herein. The invention relates to modified enzymes for de novo synthesis of polynucleotides with a desired sequence, and without the use of a template. As such, the invention provides the capability to make libraries of polynucleotides of varying sequence and varying length for research, genetic engineering, and gene therapy. Most de novo nucleic acid sequences are synthesized using solid phase phosphoramidite-techniques developed more than 30 years ago. The technique involves the sequential de-protection and synthesis of sequences built from phosphoramidite reagents corresponding to natural (or non-natural) nucleic acid bases. Phosphoramidite nucleic acid synthesis is length-limited, however, in that nucleic acids greater than 200 base pairs (bp) in length experience high rates of breakage and side reactions. Additionally, phosphoramidite synthesis produces toxic by-products, and the disposal of this waste limits the availability of nucleic acid synthesizers, and increases the costs of contract oligo production. (It is estimated that the annual demand for oligonucleotide synthesis is responsible for greater than 300,000 gallons of hazardous chemical waste, including acetonitrile, trichloroacetic acid, toluene, tetrahydrofuran, and pyridine. See LeProust et al., The invention discloses modified terminal deoxynucleotidyl transferase (TdT) enzymes that can be used for de novo synthesis of oligonucleotides in the absence of a template. Methods for creating a template-independent polymerase through a combination of computational guidance and saturation mutagenesis, with a subsequent screen to identify functional mutants, are also disclosed. Native TdT enzymes are either inefficient or completely unable to incorporate the different blocked nucleotide analogs used in template-independent synthesis schemes. The present invention provides various TdT modifications that expand the enzyme's functionality with respect to blocked nucleotide analogs, especially those with 3′-O blocking groups. In particular, modified TdTs of the invention can be used to incorporate 3′-O-Phosphate-blocked nucleotide analogs where wild type TdTs may be unable to do so. Methods of the invention include nucleic acid synthesis using 3′-O-blocked nucleotide analogs and Shrimp Alkaline Phosphatase (SAP) for controlled addition of selected nucleotides. Using enzymes and methods of the invention, it will be possible to synthesize de novo polynucleotides faster and more cheaply. As such, the invention dramatically reduces the overall cost of synthesizing custom nucleic acids. In particular, the methods can be used to create template-independent transferases that can synthesize custom oligos in a stepwise fashion using modified 3′ hydroxyl-blocked nucleotides. Because of the terminating group, synthesis pauses with the addition of each new base, whereupon the terminating group is cleaved, leaving a polynucleotide that is essentially identical to a naturally occurring nucleotide (i.e., is recognized by the enzyme as a substrate for further nucleotide incorporation). The methods and enzymes of the invention represent an important step forward in synthetic biology because the enzymes will allow for aqueous phase, template-independent oligonucleotide synthesis. Such methods represent an improvement over the prior art in that they will greatly reduce the chemical waste produced during oligonucleotide synthesis while allowing for the production of longer polynucleotides. Furthermore, because the methods replace a chemical process with a biological one, costs will be reduced, and the complexity of automated synthetic systems will also be reduced. In an embodiment, a simple five-reagent delivery system can be used to build oligonucleotides in a stepwise fashion and will enable recycling of unused reagents. The invention facilitates the synthesis of polynucleotides, such as DNA, by providing modified enzymes that can be used with nucleic acid analogs. Using the disclosed methods, a modified template-independent terminal deoxynucleotidyl transferase (TdT) is obtained that allows the enzymatically mediated synthesis of de novo oligodeoxynucleotides, thereby enabling their use in routine assembly for gene synthesis. The enzymes of the invention lend themselves to aqueous-based, enzyme-mediated methods of synthesizing polynucleotides of a predetermined sequence on a solid support. The modified enzymes of the invention will allow 3′-O-blocked dNTP analogs to be used in a step-by-step method to extend an initiating nucleic acid into a user defined sequence (see Cost savings by this approach will be achieved by exploiting the higher yield of final oligonucleotide product at a lower starting scale than currently being used as the existing industry standard (i.e., less than 1 nanomole). Future adaptation of this enzymatic approach to array based formats will allow even further and more dramatic reductions in the cost of synthesis of long oligonucleotides achievable by highly parallel synthesis. Furthermore, the enzymatic synthesis process that we propose uses only aqueous based chemistries like buffers and salts, thus greatly reducing the environmental burden of the organic waste generated by the existing phosphoramidite method. The methods of the invention may be used to modify terminal deoxynucleotidyl transferases (TdT), however other enzymes could be modified with similar methods. TdT is likely to be a successful starting enzyme because it is capable of 3′-extension activity using single strand initiating primers in a template-independent polymerization. However, prior to the invention described herein, there have been no reports of 3′-O-blocked nucleotides being incorporated into single-stranded oligonucleotide by an enzyme in the absence of a template. In fact, as Chang and Bollum reported, substitution of the 3′-hydroxyl group results in complete inactivity of available transferase enzymes. See Chang and Bollum, “Molecular Biology of Terminal Transferase, It is known that TdT can use substrates having modifications and/or substitutions at the deoxyribose sugar ring as well as the purine/pyrimidine nucleobases. For example, TdT accepts bulky modifications at the C5 of pyrimidines and the C7 of purines. See Sorensen et al., “Enzymatic Ligation of Large Biomolecules to DNA,” Native TdT is a very efficient enzyme. It has been demonstrated that TdT can polymerize extremely long homopolydeoxynucleotides of 1000 to 10,000 nucleotides in length (see Hoard et al., The distributive behavior of TdT is reinforced by Nonetheless, as described above, nucleotide synthesis with 3′-O-blocked dNTPs does not proceed with commercially-available TdT proteins. This fact is reinforced by With suitable modifications, a variety of different 3′-O-blocked dNTP analogs will be suitable for the controlled addition of nucleotides by TdT. Modified 3′-O-blocked dNTP analogs include, but are not limited to, the 3′-O-allyl, 3′-O-azidomethyl, 3′-O—NH2, 3′-O—CH2N3, 3′-O—ONHC(O)H, 3′-O—CH2SSCH3, and 3′-O—CH2CN blocking groups. Overall, the choice of the 3′-O-blocking group will be dictated by: 1) the smallest possible bulk to maximize substrate utilization by TdT, which is likely to affect kinetic uptake, and 2) the blocking group with the mildest removal conditions, preferably aqueous, and in the shortest period of time. 3′-O-blocking groups that are the suitable for use with this invention are described in WO 2003/048387; WO 2004/018497; WO 1996/023807; WO 2008/037568; Hutter D, et al. A computational model of the active site of murine TdT was created to understand the structural basis for the lack of utilization of 3′-O-blocked dNTPs by TdT. Additionally, the computer model made it possible to “fit” various modified dNTPs into the active site. The phosphate portions of the dATPs (orange) are in complex with the catalytic metal ions (green) while the alpha phosphate is positioned to be attacked by the 3′-OH of the bound oligonucleotide. The model shown in AutoDock's predicted binding mode suggests that modification to the 3′-OH will change the electrostatic interactions between two residues, Arg336 and Arg454. Although Arg336 is near the reaction center in the active site, Arg 336 is highly conserved, and early studies found that replacement of Arg336 with Gly or Ala reduced dNTP activity by 10-fold (Yang B et al. J. Mol. Biol. 1994; 269(16):11859-68). Accordingly, one motif for modification is the GGFRR motif including Arg 336 in the above structural model. Additionally, it is thought that Gly452 and Ser453 exist in a cis-peptide bond conformation (see Delarue et al., On the other hand, sequence analysis of the TdT family demonstrates a wide range of amino acids that can be accommodated at position 454. This analysis suggests structural flexibility at position 454, and surrounding residues. In another embodiment, substitutions at Arg454 to accommodate the steric bulk of a 3′-0-blocking group may require additional modifications to the α14 region to compensate for substitutions of glycine or alanine at Arg454. In other embodiments, substitutions to other residues in the all region may be required to compensate for substitution to Arg336 either instead of, or in addition to, modification of the TGSR motif. While modification to Arg336 and Arg454 may change the binding interactions of 3′-O-modified dNTPs, it may also be necessary to explore substitutions that would result in improved steric interactions of 3′-O-modified dNTPs with TdT. In order to test computationally predicted enzyme variants that show increased substrate utilization of 3′-O-blocked dNTPs, synthetic genes specifying specific amino acid substitutions were generated in appropriate plasmid vectors and introduced into cells. After expression and isolation, protein variants were screened for activity by a polymerase incorporation assay with selected 3′-O-blocked dNTP analogs. While the TGSR and GGFRR motifs are highlighted here, modifications to the flanking amino acids such as Thr331, Gly337, Lys338, Gly341, or His342 are also contemplated for providing (alone or in combination) increased incorporation of 3′-O-blocked dNTPs as discussed herein. Various in silico modeled TdT modifications capable of increased incorporation are discussed in Example 2 below. In addition to amino acid substitutions at positions 500-510 it may be necessary to delete residues to remove interference with a 3′-O-blocking group. Since these amino acids are located near the C-terminus of the protein, and exist in a relatively unstructured region, they may be deleted singly or altogether, either instead of or in combination with the modifications described above. In certain embodiments, insertion of residues into the modified TdT. For example, insertions of residues in the GGFRR or TGSR motifs or flanking regions can allow an increased rate of incorporation of 3′-O-blocked dNTP by the modified TdT. TdT modifications can include insertion of a Tyrosine residue between the Phe334 and Arg335 residues (or substitutions thereof) of the GGFRR motif. Modified TdT's of the invention include those described in As shown below, most TdTs include the GGFRR and TGSR motifs. In the following sequences, the GGFRR and TGSR motifs have been bolded and underlined for easy reference. Native calf thymus TdT is a candidate for alteration of the primary structure to achieve a suitable template-independent polymerase. However, a variety of other proteins may be explored to identify a candidate suitable for the use with 3′-O-blocked dNTP analogs, including human and murine TdT. The amino acid sequence corresponding to native calf TdT is listed in Table 1 as SEQ ID NO. 1, while the nucleic acid sequence is listed in Table 2 as SEQ ID NO. 2. In some embodiments, the resulting protein, adapted for sequence-specific de novo polynucleotide synthesis with 3′-O-modified dNTPs and NTPs, will be at least 85% identical, i.e., at least 90% identical, i.e., at least 93% identical, i.e., at least 95% identical, i.e., at least 97% identical, i.e., at least 98% identical, i.e., at least 99% identical, with SEQ ID NO. 1. Furthermore, it may be possible to truncate portions of the amino acid sequence of bovine TdT and still maintain catalytic activity. Additionally, to make isolation of recombinant proteins easier, it is common to append an N-terminal His tag sequence to the recombinant protein (see Boule J-B et al., In certain embodiments, modified enzymes of the invention may include an N-terminus truncation relative to their respective native TdT enzyme. For example, in preferred embodiments, the native enzyme may be murine TdT as provided in SEQ ID NO. 9 above. The modified TdT may be truncated at the equivalent of position 147 or 131 of the native murine TdT as shown in SEQ ID Nos. 10 and 11 respectively. Modified TdTs may include a protein tag sequence such as a His tag and additional linkers at their N-terminus as illustrated in SEQ ID Nos. 10 and 11. The His-tag portion if underlined in each of the sequences and the linker is provided in bold. Additional TdT modifications that may increase incorporation efficiency of 3′-O-blocked or other nucleotide analogs are listed in Table 10 below. While the modifications are described with referenced to the murine TdT listed in SEQ ID NO. 9, such the invention contemplates such modifications applied to the equivalent amino acids in any TdT including the truncated enzymes disclosed in SEQ ID Nos. 10 and 11 above with or without the His-tags and linkers. In various embodiments, contemplated modifications include deletion of the 5420 through E424 amino acids. Various combinations of amino acid substitutions of the invention are listed in each row 1-175 of Table 10. A variety of 3′-O-modified dNTPs and NTPs may be used with the disclosed proteins for de novo synthesis. In some embodiments, the preferred removable 3′-O-blocking group is a 3′-O-amino, a 3′-O-allyl or a 3′-O-azidomethyl. In other embodiments, the removable 3′-O-blocking moiety is selected from the group consisting of O-phenoxyacetyl; O-methoxyacetyl; O-acetyl; O-(p-toluene)-sulfonate; O-phosphate; O-nitrate; O-[4-methoxy]-tetrahydrothiopyranyl; O-tetrahydrothiopyranyl; O-[5-methyl]-tetrahydrofuranyl; O-[2-methyl,4-methoxy]-tetrahydropyranyl; O[5-methyl]-tetrahydropyranyl; and O-tetrahydrothiofuranyl (see U.S. Pat. No. 8,133,669). In other embodiments the removable blocking moiety is selected from the group consisting of esters, ethers, carbonitriles, phosphates, carbonates, carbamates, hydroxylamine, borates, nitrates, sugars, phosphoramide, phosphoramidates, phenylsulfenates, sulfates, sulfones and amino acids (see Metzker M L et al. Nuc Acids Res. 1994; 22(20):4259-67, U.S. Pat. Nos. 5,763,594, 6,232,465, 7,414,116; and 7,279,563, all of which are incorporated by reference in their entireties). Synthesis of Exemplary 3′-O-Blocked dNTP Analogs 3′-O-azidomethyl-dATP: With reference to 3′-O-azidomethyl-dTTP: Acetic acid (4.8 ml) and acetic anhydride (15.4 ml) were added to a stirred solution of 5′-O-(tertbutyldimethylsilyl)thymidine (2.0 g; 5.6 mmol) [CNH Technologies, Woburn, Mass.] in DMSO. The reaction mixture was stirred at room temperature for 48 h. A saturated NaHCO3solution (100 ml) was added, and the aqueous layer was extracted with ethyl acetate (3×100 ml). The combined organic extract was washed with a saturated solution of NaHCO3and dried over Na2SO4. After concentration, the crude product was purified by flash column chromatography (hexane/ethyl acetate) to produce 3′-O-(Methylthiomethyl)-5′-O-(tert-butyldimethylsilyl)thymidine ( 3′-O-azidomethyl-dCTP: Three and a half grams of N4-benzoyl-5′-O-(tert-butyldimethylsilyl)-2′-deoxycytidine [CNH Technologies, Woburn, Mass.] was added to 14.7 ml of DMSO to produce a 7.65 mmol solution. To this solution, acetic acid (6.7 ml) and acetic anhydride (21.6 ml) were added, and the reaction mixture was stirred at room temperature for 48 h. A saturated NaHCO3solution (100 ml) was then added and the aqueous layer was extracted with CH2Cl2(3×100 ml). The combined organic extract was washed with a saturated solution of NaHCO3and then dried over Na2SO4. After concentration, the crude product was purified by flash column chromatography (ethyl acetate/hexane) to produce N4-Benzoyl-3′-O-(methylthiomethyl)-5′-O-(tert-butyldimethylsilyl)-2′-deoxycytidine ( 3′-O-azidomethyl-dGTP: To a stirred solution of N2-isobutyryl-5′-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine (5 g; 11.0 mmol) [CNH Technologies, Woburn, Mass.] in dry DMSO (21 ml), acetic acid (10 ml) and acetic anhydride (32 ml) were added. The reaction mixture was stirred at room temperature for 48 h. A saturated NaHCO3solution (100 ml) was added and the aqueous layer was extracted with ethyl acetate (3×100 ml). The combined organic extract was washed with a saturated NaHCO3solution and dried over Na2SO4. After concentration, the crude product was purified by flash column chromatography (CH2Cl2/MeOH) to produce N2-Isobutyryl-3′-O-(methylthiomethyl)-5′-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine ( As described with respect to In preferred embodiments an enzymatic reaction is used for removal of the 3′-blocking group. Shrimp Alkaline Phosphatase (SAP) may be used in certain embodiments. SAP has one of the fastest enzymatic rates reported in the literature and has a wide range of substrate utilization. 3′-O-Methoxymethyl-dTTP: 5′-O-Benzoylthymidine (173 mg, 0.5 mmol, 1 equiv) was dissolved in 10 mL of dichloromethane under argon at ambient T. Di-isopropylethylamine (128 mg, 1 mmol, 2 equiv) was added followed by methoxymethyl bromide (124 mg, 1 mmol, 2 equiv). The mixture was stirred at ambient T for 18h. The mixture was diluted with 10 mL dichloromethane and this was washed successively with 20 mL of 5% aq HCl, and brine. The organic layer was dried with sodium sulfate and evaporated. 5′-O-Benzoyl-3′-O-methoxymethylthymidine (50 mg, 0.13 mmol) was dissolved in 5 mL of concentrated ammonium hydroxide at ambient temperature. The mixture was stirred at ambient T overnight. The mixture was diluted extracted 3 times with 10 mL portions of dichloromethane. The combined extracts were washed with brine. The organic layer was dried with sodium sulfate and evaporated. 3′-O-Methoxymethylthymidine (23 mg, 0.08 mmol) was co-evaporated with pyridine (1.5 mL×3) and dried overnight under high vacuum. The nucleoside was dissolved in a mixture of 1.5 mL of trimethylphosphate and 0.6 mL dry pyridine under Ar. The mixture was cooled in an ice bath. a first aliquot of 10 uL of POCl3was added dropwise. Five minutes later, a second aliquot of 10 uL was added. The mixture was stirred an additional 30 min. A solution of the TBA phosphate salt in dry DMF (1.25 mL) was cooled in an ice bath in a vial under Ar. This was added to the r×n mixture dropwise over 10 sec. Immediately the pre-weighed solid proton sponge (21 mg, 1.25 equiv) was added as a solid in one portion. The mixture was stirred for 25 min after this addition and was quenched with 5 mL of cold TEAB buffer. The mixture was stirred in the ice bath for 10 min and then transferred to a small RB flask for FPLC separation. Final separation was accomplished by reverse phase HPLC using a water/acetonitrile gradient containing 0.1 mM formic acid. 3′-O-Methylthiomethyl-dCTP: To a suspension of deoxycytidine (1 g, 4.4 mmol) in 25 mL of methanol was added N,N-dimethylformamide dimethyl acetal (1.75 mL, 13.2 mmol). The mixture was stirred overnight at ambient temperature. The reaction mixture was evaporated, and the residue was purified by flash chromatography using a DCM/methanol gradient as eluant. N6-Formamidino-5′-O-benzoyldeoxy-3′-O-methylthiomethyldeoxycytidine (250 mg, 0.41 mmol) was dissolved in 10 mL of methanol and 10 mL conc aqueous ammonium hydroxide. The mixture was stirred at ambient temperature for 18 h and then evaporated under reduced pressure. The residue was purified by column chromatography (DCM/Methanol 98:2 to 90:10) to afford 170 mg (93%) of the desired nucleoside as a slightly yellow solid. 3′-O-Methylthiomethyl dexoxycytidine (25.0 mg, 0.09 mmol) in a 25 mL vial was co-evaporated with anhydrous pyridine (3×1 mL) and dried over the weekend. Trimethyl phosphate (0.7 mL) was added to dissolve the nucleoside and cooled in an ice bath to 0° C. Phosphoryl chloride (28 μL, 0.3 mmol) was added slowly (12 μL, 5 min later 8 μL, 30 min later 8 μL) and the reaction was stirred for 2 h at 0° C. The di(tetrabutylammonium) hydrogen pyrophosphate was dissolved in anhydrous DMF (1 mL), this mixture was cooled to 0° C. and added to the reaction mixture. Proton sponge (9.2 mg, 0.04 mmol) was added and the reaction was stirred at 0° C. for 2 h. To the reaction mixture was added 1 M triethylammonium bicarbonate buffer (TEAB) (2 mL) and the mixture was stirred for 1 h. The mixture was then transferred to round-bottom flask, 50 mL×3 of miliQ water was added and mixture was concentrated to dryness. The residue was dissolved in miliQ water (11 mL) and loaded onto an AKTA FPLC at room temperature. The fractions containing the triphosphate (F48-F52) were evaporated under reduced pressure at 40° C., and the residue was then lyophilized. The triphosphate was dried to afford the desired triphosphate (12 mg, 16.5%). Murine (mur) TdT variants originated from 380 aa synthetic gene. This backbone is a truncated version of WT murine TdT and represents a catalytic core of the ET sequence. Chemically synthesized TdT constructs were cloned into a pRSET A bacterial expression vector, featuring an N-terminal 6×-histidine tag and enterokinase cleavage site (ThermoFisher Scientific GeneArt Gene Synthesis). Synthetic TdT plasmids were maintained in DH5alpha cells (Biopioneer) plated on LB agar plates containing 100 ug/ml carbenicillin. For expression, the pRSETA-murine TdT plasmids were transformed into BL21 (DE3) pLysS cells (Thermo-Fisher) by incubating plasmids and cells on ice for 20 min., followed by a 30 sec. heat shock at 42° C., followed by addition of SOC media and incubation with shaking at 37° C. for 30-60 min. After addition of SOC media to cells, the entire volume (typically 60 ul) were plated on LB agar plates containing 100 ug/mL carbenicillin plus 34 ug/mL chloramphenicol. Cells from 10 mL cultures (24-well plates, Corning) were harvested by centrifugation (3000×g, 15 min), then lysed in B-PER lysis buffer (Thermo-Fisher) containing lysozyme, protease inhibitors, and 100 mM NaCl. Pellets were soaked 1×60 min. in TBS buffer and supernatants collected for purification. The supernatant was bound onto 50 uL Ni-NTA bead (GE Life Sciences) slurry in 24-well plates for 30 min. The bead slurry was then washed 3×50 mM Tris-HCl, pH 8, 500 mM NaCl (500 uL), followed by washing 4×50 mM Tris-HCl, pH 8, 500 mM NaCl, 50 mM Imidazole (200 uL). The protein was then recovered by treating with 50 mM Tris-HCl, pH 8, 500 mM NaCl, 300 mM Imidazole (50 uL), then 50 mM Tris-HCl, pH 8, 500 mM NaCl, 300 mM Imidazole (130 uL), and finally 50 mM Tris-HCl, pH 8, 500 mM NaCl, 1M Imidazole (50 uL). Recovered fractions were analyzed by taking 2.5 ul sample and running on 8% NuPage gel (Thermo-Fisher), 200 V for 50 min, denaturing conditions. Gel stained with Coomassie Blue. The eluted protein was buffer exchanged using a 7.5 MWCO desalting column (Thermo-Fisher) and sored at −80° C. (Storage Buffer=20 mM Tris-HCl, pH 6.8, 50 mM NaOAc; 0.01% Triton X-100 and 10% Glycerol). TdT activity screening was performed via a dNTP polymerase extension reaction using different 3′-O-blocked dNTP analogs and a biotinylated oligonucleotide: Reactions were typically set up in a 96 well plate. Reactions were performed by making a master mix with final concentrations of the following components: 0.2 U PPase (Thermo-Fisher), 10 pmol of oligonucleotide, 75 uM dNTP (see below), 1×TdT reaction buffer (5× from Thermo-Fisher) to a final volume of 10 ul. Reactions were initiated by adding a defined volume (typically 2 ul) of TdT variants in different wells and incubating the reaction mix at 37° C. for 5 min and 60 min time points. Reactions were terminated by removal of a 10 ul aliquot and adding to 5 ul of 250 mM EDTA. dNTPs Tested: Biotinylated oligos in the quenched reaction mix were bound to Streptavidin beads (0.77 um, Spherotech). The beads were then transferred to filter plates (Pall Corporation) and washed several times with water. The oligonucleotides were cleaved from the solid support by incubating the plate with cleavage buffer (10% Diisopropyl-amine in methanol) at 50° C. for 30 min followed by elution in water. The eluted samples were dried and dissolved in 30 μl of water containing oligonucleotide sizing standards (two oligonucleotides (ChemGenes Corporation) that are approximately 15-20 bases smaller or larger than the starting 42-mer oligonucleotide). Oligonucleotides were then analyzed for extension efficiency by Capillary Gel Electrophoresis (Oligo Pro II, Advanced Analytical Technologies Inc.). Several amino acid modifications to the GGFRR and TGSR motifs and flanking amino acids discussed above were modeled in silico to determine modifications capable of increased incorporation of 3′-O-blocked dNTP analogs as described above. Single, double, and triple amino acid substitutions as well amino acid insertions were modeled. Table 11 below shows modifications found to elicit increased incorporation. Amino acid positions are provided with reference to murine TdT but are applicable to conserved sequences of any TdT. Rows in Table 11 describe a base modification to one or more amino acids in or flanking the GGFRR motif. Columns include additional combinations of modifications to other amino acids such as those in and flanking the TGSR motif. DNA and the nucleotides that comprise DNA are highly negatively charged due to the phosphate groups within the nucleotides. See Lipfert J, Doniach S, Das R, Herschlag D. Understanding Nucleic Acid-Ion Interactions, Annu Rev Biochem. 2014; 83: 813-841, incorporated herein by reference. 3′-PO4-dNTPs have an even greater negative charge relative to natural nucleotides due to the additional phosphate group at the 3′-position. The increased negative charge may affect the ability of the TdT to incorporate the modified nucleotides. In certain embodiments, engineered TdT enzymes of the invention may be modified for efficient incorporation of 3′-phosphate-dNTPs by neutralizing the negative charges with positive charges on the modified TdT. The Average number of Neighboring Atoms Per Sidechain Atom (AvNAPSA) algorithm within the Rosetta protein software suite3 was used to identify mutations that will increase the positive charge in and around the enzymatic active site of TdT. By increasing a key parameter of the AvNAPSA algorithm, termed surface atom cutoff, sequence positions in the active site of TdT were targeted. The surface charge of proteins was manipulated by mutating solvent-exposed polar residues to charged residues, with the amount of solvent exposure determined by the number of neighboring non-self atoms. See, Miklos A E, et al., Structure-Based Design of Supercharged, Highly Thermoresistant Antibodies, Chemistry & Biology, Volume 19, Issue 4, 20 Apr. 2012, Pages 449-455; Kaufmann K W, et al., Practically useful: what the Rosetta protein modeling suite can do for you, Biochemistry. 2010 Apr. 13; 49(14):2987-98; the content of each of which is incorporate herein by reference. Increasing the surface_atom_cutoff term allows AvNAPSA to consider sequence positions with a higher number of neighboring atoms, such as positions within an enzyme active site. A summary of positions identified in TdT using AvNAPSA as being potentially useful for more efficient incorporation of 3′-phosphate-dNTP is shown in Table 12. References and citations to other documents, such as patents, patent applications, patent publications, journals, books, papers, web contents, have been made throughout this disclosure. All such documents are hereby incorporated herein by reference in their entirety for all purposes. Various modifications of the invention and many further embodiments thereof, in addition to those shown and described herein, will become apparent to those skilled in the art from the full contents of this document, including references to the scientific and patent literature cited herein. The subject matter herein contains important information, exemplification and guidance that can be adapted to the practice of this invention in its various embodiments and equivalents thereof. The invention includes methods for identifying polymerases, such as modified terminal nucleotidyl transferases (TdT), that are capable of binding nucleotides comprising removable 3′-O-blocking moieties to a nucleic acid initiator, without the use of a template. The invention further includes the identified polymerases, and methods of using the polymerases for de novo synthesis of predetermined oligonucleotide sequences. 1. A modified terminal deoxynucleotidyl transferase (TdT) comprising a mutation selected from the group consisting of E33K, E180L, E180K, M192E, M192K, M192W, W303H, L381K, L381Q, L381R, L381V, W450H, R454I, R454T, R454K, E457K, R461V, R461Q, R461V, N474R, and N474K, said modified TdT capable of adding a nucleotide analog comprising a removable blocking moiety at a 3′-Oxygen of the analog to a 3′-OH of a nucleic acid initiator in the absence of a nucleic acid template. 2. The modified TdT of 3. The modified TdT of 4. The modified TdT of 5. The modified TdT of 6. The modified TdT of 7. The modified TdT of 8. The modified TdT of 9. The modified TdT of 10. The modified TdT of 11. The modified TdT of 12. The modified TdT of 13. The modified TdT of 14. The modified TdT of 15. The modified TdT of 16. The modified TdT of 17. The modified TdT of 18. The modified TdT of 19. The modified TdT of 20. The modified TdT of 21. The modified TdT of 22. The modified TdT of 23. The modified TdT of 24. The modified TdT of 25. The modified TdT of 26. The modified TdT of 27. The modified TdT of RELATED APPLICATIONS
FIELD OF THE INVENTION
BACKGROUND
SUMMARY
BRIEF DESCRIPTION OF THE DRAWINGS
DESCRIPTION OF THE INVENTION
MAQQRQHQRL PMDPLCTASS GPRKKRPRQV GASMASPPHD IKFQNLVLFI LEKKMGTTRR NFLMELARRK GFRVENELSD SVTHIVAENN SGSEVLEWLQ VQNIRASSQL ELLDVSWLIE SMGAGKPVEI TGKHQLVVRT DYSATPNPGF QKTPPLAVKK ISQYACQRKT TLNNYNHIFT DAFEILAENS EFKENEVSYV TFMRAASVLK SLPFTIISMK DTEGIPCLGD KVKCIIEEII EDGESSEVKA VLNDERYQSF KLFTSVFGVG LKTSEKWFRM GFRSLSKIMS DKTLKFTKMQ KAGFLYYEDL VSCVTRAEAE AVGVLVKEAV WAFLPDAFVT MTGGFRRGKK IGHDVDFLIT SPGSAEDEEQ LLPKVINLWE KKGLLLYYDL VESTFEKFKL PSRQVDTLDH FQKCFLILKL HHQRVDSSKS NQQEGKTWKA IRVDLVMCPY ENRAFALLGW TGSRQFERDI RRYATHERKM MLDNHALYDK TKRVFLKAES EEEIFAHLGL DYIEPWERNA ctcttctgga gataccactt gatggcacag cagaggcagc atcagcgtct tcccatggat ccgctgtgca cagcctcctc aggccctcgg aagaagagac ccaggcaggt gggtgcctca atggcctccc ctcctcatga catcaagttt caaaatttgg tcctcttcat tttggagaag aaaatgggaa ccacccgcag aaacttcctc atggagctgg ctcgaaggaa aggtttcagg gttgaaaatg agctcagtga ttctgtcacc cacattgtag cagaaaacaa ctctggttca gaggttctcg agtggcttca ggtacagaac ataagagcca gctcgcagct agaactcctt gatgtctcct ggctgatcga aagtatggga gcaggaaaac cagtggagat tacaggaaaa caccagcttg ttgtgagaac agactattca gctaccccaa acccaggctt ccagaagact ccaccacttg ctgtaaaaaa gatctcccag tacgcgtgtc aaagaaaaac cactttgaac aactataacc acatattcac ggatgccttt gagatactgg ctgaaaattc tgagtttaaa gaaaatgaag tctcttatgt gacatttatg agagcagctt ctgtacttaa atctctgcca ttcacaatca tcagtatgaa ggatacagaa ggaattccct gcctggggga caaggtgaag tgtatcatag aggaaattat tgaagatgga gaaagttctg aagttaaagc tgtgttaaat gatgaacgat atcagtcctt caaactcttt acttctgttt ttggagtggg actgaagaca tctgagaaat ggttcaggat ggggttcaga tctctgagta aaataatgtc agacaaaacc ctgaaattca caaaaatgca gaaagcagga tttctctatt atgaagacct tgtcagctgc gtgaccaggg ccgaagcaga ggcggttggc gtgctggtta aagaggctgt gtgggcattt ctgccggatg cctttgtcac catgacagga ggattccgca ggggtaagaa gattgggcat gatgtagatt ttttaattac cagcccagga tcagcagagg atgaagagca acttttgcct aaagtgataa acttatggga aaaaaaggga ttacttttat attatgacct tgtggagtca acatttgaaa agttcaagtt gccaagcagg caggtggata ctttagatca ttttcaaaaa tgctttctga ttttaaaatt gcaccatcag agagtagaca gtagcaagtc caaccagcag gaaggaaaga cctggaaggc catccgtgtg gacctggtta tgtgccccta cgagaaccgt gcctttgccc tgctaggctg gactggctcc cggcagtttg agagagacat ccggcgctat gccacacacg agcggaagat gatgctggat aaccacgctt tatatgacaa gaccaagagg gtatttctca aagcggaaag tgaagaagaa atctttgcac atctgggatt ggactacatt gaaccatggg aaagaaatgc ttaggagaaa gctgtcaact tttttctttt ctgttctttt tttcaggtta gacaaattat gcttcatatt ataatgaaag atgccttagt caagtttggg attctttaca ttttaccaag atgtagattg cttctagaaa taagtagttt tggaaacgtg atcaggcacc ccctgggtta tgctctggca agccatttgc aggactgatg tgtagaactc gcaatgcatt ttccatagaa acagtgttgg aattggtggc tcatttccag ggaagttcat caaagcccac tttgcccaca gtgtagctga aatactgtat acttgccaat aaaaatagga aac Met Arg Gly Ser His His His His His His Arg Thr Asp Tyr Ser Ala Thr Pro Asn Pro Gly Phe Gln Lys Thr Pro Pro Leu Ala Val Lys Lys Ile Ser Gln Tyr Ala Cys Gln Arg Lys Thr Thr Leu Asn Asn Tyr Asn His Ile Asp Ala Phe Glu Ile Leu Ala Glu Asn Ser Glu Phe Lys Glu Asn Glu Val Ser Tyr Val Thr Phe Met Arg Ala Ala Ser Val Leu Lys Ser Leu Pro Phe Thr Ile Ile Ser Met Lys Asp Thr Phe Thr Glu Gly Ile Pro Cys Leu Gly Asp Lys Val Lys Cys Ile Ile Glu Glu Ile Ile Glu Asp Gly Glu Ser Ser Glu Val Lys Ala Val Leu Asn Asp Glu Arg Tyr Gln Ser Phe Lys Leu Ser Val Phe Gly Val Gly Leu Lys Thr Ser Glu Lys Trp Phe Arg Met Gly Phe Thr Phe Arg Ser Leu Ser Lys Ile Met Ser Asp Lys Thr Leu Lys Lys Met Gln Lys Ala Gly Phe Leu Tyr Tyr Glu Asp Leu Val Ser Cys Val Thr Arg Ala Glu Ala Glu Ala Val Gly Val Leu Val Lys Glu Ala Val Trp Ala Phe Leu Pro Asp Ala Phe Val Thr Met Thr Gly Gly Phe Arg Arg Gly Lys Lys Ile Gly His Asp Val Asp Phe Leu Ile Thr Ser Pro Gly Ser Ala Glu Asp Glu Glu Gln Leu Leu Pro Lys Val Ile Asn Leu Trp Glu Lys Lys Gly Leu Leu Leu Tyr Tyr Asp Leu Val Glu Ser Thr Phe Glu Lys Phe Lys Phe Thr Leu Pro Ser Arg Gln Val Asp Thr Leu Asp His Phe Gln Lys Cys Phe Leu Ile Leu Lys Leu His His Gln Arg Val Asp Ser Ser Lys Ser Asn Gln Gln Glu Gly Lys Thr Trp Lys Ala Ile Arg Val Asp Leu Val Met Cys Pro Tyr Glu Asn Arg Ala Phe Ala Leu Leu Gly Trp Thr Gly Ser Arg Gln Phe Glu Arg Asp Ile Arg Arg Tyr Ala Thr His Glu Arg Lys Met Met Leu Asp Asn His Ala Leu Tyr Asp Lys Thr Lys Arg Val Phe Leu Lys Ala Glu Ser Glu Glu Glu Ile Phe Ala His Leu Gly Leu Asp Tyr Ile Glu Pro Trp Glu Arg Asn Ala atgagaggat cgcatcacca tcaccatcac agaacagact attcagctac cccaaaccca ggcttccaga agactccacc acttgctgta aaaaagatct cccagtacgc gtgtcaaaga aaaaccactt tgaacaacta taaccacata ttcacggatg cctttgagat actggctgaa aattctgagt ttaaagaaaa tgaagtctct tatgtgacat ttatgagagc agcttctgta cttaaatctc tgccattcac aatcatcagt atgaaggata cagaaggaat tccctgcctg ggggacaagg tgaagtgtat catagaggaa attattgaag atggagaaag ttctgaagtt aaagctgtgt taaatgatga acgatatcag tccttcaaac tctttacttc tgtttttgga gtgggactga agacatctga gaaatggttc aggatggggt tcagatctct gagtaaaata atgtcagaca aaaccctgaa attcacaaaa atgcagaaag caggatttct ctattatgaa gaccttgtca gctgcgtgac cagggccgaa gcagaggcgg ttggcgtgct ggttaaagag gctgtgtggg catttctgcc ggatgccttt gtcaccatga caggaggatt ccgcaggggt aagaagattg ggcatgatgt agatttttta attaccagcc caggatcagc agaggatgaa gagcaacttt tgcctaaagt gataaactta tgggaaaaaa agggattact tttatattat gaccttgtgg agtcaacatt tgaaaagttc aagttgccaa gcaggcaggt ggatacttta gatcattttc aaaaatgctt tctgatttta aaattgcacc atcagagagt agacagtagc aagtccaacc agcaggaagg aaagacctgg aaggccatcc gtgtggacct ggttatgtgc ccctacgaga accgtgcctt tgccctgcta ggctggactg gctcccggca gtttgagaga gacatccggc gctatgccac acacgagcgg aagatgatgc tggataacca cgctttatat gacaagacca agagggtatt tctcaaagcg gaaagtgaag aagaaatctt tgcacatctg ggattggact acattgaacc atgggaaaga aatgcttaag cttgcgc Met Arg Gly Ser His His His His His His Lys Thr Pro Pro Leu Ala Val Lys Lys Ile Ser Gln Tyr Ala Cys Gln Arg Lys Thr Thr Leu Asn Asn Tyr Asn His Ile Asp Ala Phe Glu Ile Leu Ala Glu Asn Ser Glu Phe Lys Glu Asn Glu Val Ser Tyr Val Thr Phe Met Arg Ala Ala Ser Val Leu Lys Ser Leu Pro Phe Thr Ile Ile Ser Met Lys Asp Thr Phe Thr Glu Gly Ile Pro Cys Leu Gly Asp Lys Val Lys Cys Ile Ile Glu Glu Ile Ile Glu Asp Gly Glu Ser Ser Glu Val Lys Ala Val Leu Asn Asp Glu Arg Tyr Gln Ser Phe Lys Leu Ser Val Phe Gly Val Gly Leu Lys Thr Ser Glu Lys Trp Phe Arg Met Gly Phe Thr Phe Arg Ser Leu Ser Lys Ile Met Ser Asp Lys Thr Leu Lys Lys Met Gln Lys Ala Gly Phe Leu Tyr Tyr Glu Asp Leu Val Ser Cys Val Thr Arg Ala Glu Ala Glu Ala Val Gly Val Leu Val Lys Glu Ala Val Trp Ala Phe Leu Pro Asp Ala Phe Val Thr Met Thr Gly Gly Phe Arg Arg Gly Lys Lys Ile Gly His Asp Val Asp Phe Leu Ile Thr Ser Pro Gly Ser Ala Glu Asp Glu Glu Gln Leu Leu Pro Lys Val Ile Asn Leu Trp Glu Lys Lys Gly Leu Leu Leu Tyr Tyr Asp Leu Val Glu Ser Thr Phe Glu Lys Phe Lys Phe Thr Leu Pro Ser Arg Gln Val Asp Thr Leu Asp His Phe Gln Lys Cys Phe Leu Ile Leu Lys Leu His His Gln Arg Val Asp Ser Ser Lys Ser Asn Gln Gln Glu Gly Lys Thr Trp Lys Ala Ile Arg Val Asp Leu Val Met Cys Pro Tyr Glu Asn Arg Ala Phe Ala Leu Leu Gly Trp Thr Gly Ser Arg Gln Phe Glu Arg Asp Ile Arg Arg Tyr Ala Thr His Glu Arg Lys Met Met Leu Asp Asn His Ala Leu Tyr Asp Lys Thr Lys Arg Val Phe Leu Lys Ala Glu Ser Glu Glu Glu Ile Phe Ala His Leu Gly Leu Asp Tyr Ile Glu Pro Trp Glu Arg Asn Ala atgagaggat cgcatcacca tcaccatcac aagactccac cacttgctgt aaaaaagatc tcccagtacg cgtgtcaaag aaaaaccact ttgaacaact ataaccacat attcacggat gcctttgaga tactggctga aaattctgag tttaaagaaa atgaagtctc ttatgtgaca tttatgagag cagcttctgt acttaaatct ctgccattca caatcatcag tatgaaggat acagaaggaa ttccctgcct gggggacaag gtgaagtgta tcatagagga aattattgaa gatggagaaa gttctgaagt taaagctgtg ttaaatgatg aacgatatca gtccttcaaa ctctttactt ctgtttttgg agtgggactg aagacatctg agaaatggtt caggatgggg ttcagatctc tgagtaaaat aatgtcagac aaaaccctga aattcacaaa aatgcagaaa gcaggatttc tctattatga agaccttgtc agctgcgtga ccagggccga agcagaggcg gttggcgtgc tggttaaaga ggctgtgtgg gcatttctgc cggatgcctt tgtcaccatg acaggaggat tccgcagggg taagaagatt gggcatgatg tagatttttt aattaccagc ccaggatcag cagaggatga agagcaactt ttgcctaaag tgataaactt atgggaaaaa aagggattac ttttatatta tgaccttgtg gagtcaacat ttgaaaagtt caagttgcca agcaggcagg tggatacttt agatcatttt caaaaatgct ttctgatttt aaaattgcac catcagagag tagacagtag caagtccaac cagcaggaag gaaagacctg gaaggccatc cgtgtggacc tggttatgtg cccctacgag aaccgtgcct ttgccctgct aggctggact ggctcccggc agtttgagag agacatccgg cgctatgcca cacacgagcg gaagatgatg ctggataacc acgctttata tgacaagacc aagagggtat ttctcaaagc ggaaagtgaa gaagaaatct ttgcacatct gggattggac tacattgaac catgggaaag aaatgcttaa gcttgcgc Met Arg Gly Ser His His His His His His Ile Ser Gln Tyr Ala Cys Gln Arg Lys Thr Thr Leu Asn Asn Tyr Asn His Ile Asp Ala Phe Glu Ile Leu Ala Glu Asn Ser Glu Phe Lys Glu Asn Glu Val Ser Tyr Val Thr Phe Met Arg Ala Ala Ser Val Leu Lys Ser Leu Pro Phe Thr Ile Ile Ser Met Lys Asp Thr Phe Thr Glu Gly Ile Pro Cys Leu Gly Asp Lys Val Lys Cys Ile Ile Glu Glu Ile Ile Glu Asp Gly Glu Ser Ser Glu Val Lys Ala Val Leu Asn Asp Glu Arg Tyr Gln Ser Phe Lys Leu Ser Val Phe Gly Val Gly Leu Lys Thr Ser Glu Lys Trp Phe Arg Met Gly Phe Thr Phe Arg Ser Leu Ser Lys Ile Met Ser Asp Lys Thr Leu Lys Lys Met Gln Lys Ala Gly Phe Leu Tyr Tyr Glu Asp Leu Val Ser Cys Val Thr Arg Ala Glu Ala Glu Ala Val Gly Val Leu Val Lys Glu Ala Val Trp Ala Phe Leu Pro Asp Ala Phe Val Thr Met Thr Gly Gly Phe Arg Arg Gly Lys Lys Ile Gly His Asp Val Asp Phe Leu Ile Thr Ser Pro Gly Ser Ala Glu Asp Glu Glu Gln Leu Leu Pro Lys Val Ile Asn Leu Trp Glu Lys Lys Gly Leu Leu Leu Tyr Tyr Asp Leu Val Glu Ser Thr Phe Glu Lys Phe Lys Phe Thr Leu Pro Ser Arg Gln Val Asp Thr Leu Asp His Phe Gln Lys Cys Phe Leu Ile Leu Lys Leu His His Gln Arg Val Asp Ser Ser Lys Ser Asn Gln Gln Glu Gly Lys Thr Trp Lys Ala Ile Arg Val Asp Leu Val Met Cys Pro Tyr Glu Asn Arg Ala Phe Ala Leu Leu Gly Trp Thr Gly Ser Arg Gln Phe Glu Arg Asp Ile Arg Arg Tyr Ala Thr His Glu Arg Lys Met Met Leu Asp Asn His Ala Leu Tyr Asp Lys Thr Lys Arg Val Phe Leu Lys Ala Glu Ser Glu Glu Glu Ile Phe Ala His Leu Gly Leu Asp Tyr Ile Glu Pro Trp Glu Arg Asn Ala atgagaggat cgcatcacca tcaccatcac atctcccagt acgcgtgtca aagaaaaacc actttgaaca actataacca catattcacg gatgcctttg agatactggc tgaaaattct gagtttaaag aaaatgaagt ctcttatgtg acatttatga gagcagcttc tgtacttaaa tctctgccat tcacaatcat cagtatgaag gatacagaag gaattccctg cctgggggac aaggtgaagt gtatcataga ggaaattatt gaagatggag aaagttctga agttaaagct gtgttaaatg atgaacgata tcagtccttc aaactcttta cttctgtttt tggagtggga ctgaagacat ctgagaaatg gttcaggatg gggttcagat ctctgagtaa aataatgtca gacaaaaccc tgaaattcac aaaaatgcag aaagcaggat ttctctatta tgaagacctt gtcagctgcg tgaccagggc cgaagcagag gcggttggcg tgctggttaa agaggctgtg tgggcatttc tgccggatgc ctttgtcacc atgacaggag gattccgcag gggtaagaag attgggcatg atgtagattt tttaattacc agcccaggat cagcagagga tgaagagcaa cttttgccta aagtgataaa cttatgggaa aaaaagggat tacttttata ttatgacctt gtggagtcaa catttgaaaa gttcaagttg ccaagcaggc aggtggatac tttagatcat tttcaaaaat gctttctgat tttaaaattg caccatcaga gagtagacag tagcaagtcc aaccagcagg aaggaaagac ctggaaggcc atccgtgtgg acctggttat gtgcccctac gagaaccgtg cctttgccct gctaggctgg actggctccc ggcagtttga gagagacatc cggcgctatg ccacacacga gcggaagatg atgctggata accacgcttt atatgacaag accaagaggg tatttctcaa agcggaaagt gaagaagaaa tctttgcaca tctgggattg gactacattg aaccatggga aagaaatgct taagcttgcg c MDPLQAVHLG PRKKRPRQLG TPVASTPYDI RFRDLVLFIL EKKMGTTRRA FLMELARRKG FRVENELSDS VTHIVAENNS GSDVLEWLQL QNIKASSELE LLDISWLIEC MGAGKPVEMM GRHQLVVNRN SSPSPVPGSQ NVPAPAVKKI SQYACQRRTT LNNYNQLFTD ALDILAENDE LRENEGSCLA FMRASSVLKS LPFPITSMKD TEGIPCLGDK VKSIIEGIIE DGESSEAKAV LNDERYKSFK LFTSVFGVGL KTAEKWFRMG FRTLSKIQSD KSLRFTQMQK AGFLYYEDLV SCVNRPEAEA VSMLVKEAVV TFLPDALVTM TGGFRRGKMT GHDVDFLITS PEATEDEEQQ LLHKVTDFWK QQGLLLYCDI LESTFEKFKQ PSRKVDALDH FQKCFLILKL DHGRVHSEKS GQQEGKGWKA IRVDLVMCPY DRRAFALLGW TGSRQFERDL RRYATHERKM MLDNHALYDR TKRVFLEAES EEEIFAHLGL DYIEPWERNA SEQ ID No. 10: Murine del-147 with His-tag and linker MRGSHHHHHHGMASMTGGQQMGRDLYDDDDKDRWGSELEKKISQYACQRR TTLNNYNQLFTDALDILAENDELRENEGSCLAFMRASSVLKSLPFPITSM KDTEGIPCLGDKVKSIIEGIIEDGESSEAKAVLNDERYKSFKLFTSVFGV GLKTAEKWFRMGFRTLSKIQSDKSLRFTQMQKAGFLYYEDLVSCVNRPEA EAVSMLVKEAVVTFLPDALVTMTGGFRRGKMTGHDVDFLITSPEATEDEE QQLLHKVTDFWKQQGLLLYCDILESTFEKFKQPSRKVDALDHFQKCFLIL KLDHGRVHSEKSGQQEGKGWKAIRVDLVMCPYDRRAFALLGWTGSRQFER DLRRYATHERKMMLDNHALYDRTKRVFLEAESEEEIFAHLGLDYIEPWER NA SEQ ID No. 11: Murine del-131 with His-tag and linker MRGSHHHHHHGMASMTGGQQMGRENLYFQGSPSPVPGSQNVPAPAVKKIS QYACQRRTTLNNYNQLFTDALDILAENDELRENEGSCLAFMRASSVLKSL PFPITSMKDTEGIPCLGDKVKSIIEGIIEDGESSEAKAVLNDERYKSFKL FTSVFGVGLKTAEKWFRMGFRTLSKIQSDKSLRFTQMQKAGFLYYEDLVS CVNRPEAEAVSMLVKEAVVTFLPDALVTMTGGFRRGKMTGHDVDFLITSP EATEDEEQQLLHKVTDFWKQQGLLLYCDILESTFEKFKQPSRKVDALDHF QKCFLILKLDHGRVHSEKSGQQEGKGWKAIRVDLVMCPYDRRAFALLGWT GSRQFERDLRRYATHERKMMLDNHALYDRTKRVFLEAESEEEIFAHLGLD YIEPWERNA 1 A446S 2 A446T W450H 3 A446T 4 A510G 5 E177D E180D 6 E177D 7 E177K E180K R454A 8 E177K E180K 9 E177K 10 E177S 11 E180C 12 E180D E177D W450H 13 E180D L189M M192E L381K 14 E180D L189M M192E L381K W450H R454A R461Q 15 E180D M192E L381K R454T R461Q 16 E180D M192E L381Q R454K N474A 17 E180D M192E R454K 18 E180D M192K L381K R454K R461Q N474R 19 E180D M192K L381Q R454T N474K 20 E180D W450Y 21 E180G 22 E180K L381K W450H R454A N474A 23 E180K L381Q W450H R461V 24 E180K M192E L381K R454T N474K 25 E180K M192E L381A W450H R454T R461V 26 E180K M192E L381K W450H R454I R461Q N474R 27 E180K M192E L381V N474A 28 E180K M192E L381W R454I R461V 29 E180K M192E R4541 30 E180K M192E R454T 31 E180K M192K G337D L381R R454I N474K 32 E180K M192K L381A R454A R461Q N474R 33 E180K M192K L381A R454K N474K 34 E180K M192K L381K R454K N474R 35 E180K M192K L381K R454T N474K 36 E180K M192K L381K W450H R454I N474R 37 E180K M192K L381R W450H R461V N474R 38 E180K M192K R454I 39 E180K M192K R454K R461V N474R 40 E180K M192P R454T 41 E180K M192W L381A R454I R461Q N474K 42 E180K M192W L381K R454K N474A 43 E180K M192W L381R W450H 44 E180K M192W L381R W450H R454K R461Q 45 E180K M192W L381V R454A 46 E180K M192W R454T R461Q 47 E180K R335K 48 E180K R454A 49 E180K R454I R461V 50 E180K R454K 51 E180K R454T 52 E180L E226D L381Q R454A R461V N474A 53 E180L L381A R454A R461Q N474K 54 E180L L381A R454I R461Q 55 E180L L381A R454I R461Q N474K 56 E180L M192E L381K R461Q N474K 57 E180L M192E L381K R461Q N474K 58 E180L M192K L381K R454T N474A 59 E180L W450H R454T R461Q 60 E33K R307T F187Y 61 F405R 62 F405Y N474R W450H 63 F405Y 64 K403S 65 L347H 66 L381I N474R 67 L381K R454K 68 L381Q E180K N474R 69 L381Q E180K 70 L381Q W450H 71 L381Q 72 L381R E180K N474R 73 L381R E180K 74 L381R N474R 75 L381R 76 L381V E180K 77 L381V N474R 78 L381V 79 L381W N474R 80 L381W R454T R461V N474R 81 L381Y W450H 82 L398F E180K N474R 83 L398F E180K 84 L398F N474R 85 L398H E180K N474R 86 L398M E180K N474R 87 L398M F405Y 88 L398M N474R 89 L398M W450H 90 L472F G449A N474R R454D 91 L472F N474R R454D E457A 92 L472F N474R R454K E457D 93 L472F N474R R454Q E457D 94 L472F N474R R454Q E457S 95 L472F R454K E457D R461A 96 M192 97 M192A 98 M192E L381R R454T R461V N474A 99 M192E L381V R454I R461V 100 M192E L381V R454I R461V N474K 101 M192E L381V W450H R454K 102 M192E L381V W450H R454K R461V N474A 103 M192E R454A 104 M192G 105 M192H 106 M192K L381Q R454K N474R 107 M192K L381Q R461Q N474K 108 M192W L381R R454K N474K 109 Q390R 110 Q455A R454G 111 Q455E 112 Q455F 113 Q455H 114 Q455L 115 Q455M 116 Q455N R454G 117 Q455S R454G W450H 118 Q455T R454G 119 Q455T 120 R336N H342R 121 R454T G337H 122 R454T G341C 123 R432Q D434H R336Q H342R 124 R454C 125 R454E 126 R454G Q455R 127 R454H W450H 128 R454H W450Y 129 R454H 130 R454I 131 R454M 132 R454N 133 R454P 134 R454Q 135 R454S 136 R454T T331A 137 R454T 138 R454V 139 R461K 140 S453A R454A 141 S453G R454A W450H 142 S453G W450H 143 S453T W450H 144 S453T 145 T451S 146 T455V 147 T455Y 148 E457K 149 V436A W450H 150 V436A 151 L381Q W450H 152 E33K W303H 153 E180K L381R 154 N304K 155 N304R 156 N509K 157 N509R 158 D434K 159 D434R 160 D170K 161 D170R 162 D173K 163 D173R 164 E457K 165 E457R 166 D473K 167 D473R 168 Q402K 169 Q402R 170 D399K 171 D339R 172 E382K 173 E382R 174 Q455K 175 Q455R EXAMPLES
Example 1: Protein Modifications
Activity Screens:
SEQ ID NO. 12 5BiosG/TAATAATAATAATAATAATAATAATAATAATAATAATTTTTT (ChemGenes Corporation) 3’-O-azidomethyl-dTTP see description above 3’-O-azidomethyl-dATP see description above 3’-O-azidomethyl-dGTP see description above 3’-O-MOM-dTTP see description above 3’-O-MTM-dCTP see description above 3’-aminoxy-dTTP Firebird BioMolecular Sciences LLC 3’-aminoxy-dATP Firebird BioMolecular Sciences LLC 3’-aminoxy-dGTP Firebird BioMolecular Sciences LLC 3’-O-methyl-dATP TriLink BioTechnologies LLC 3’-O-methyl-dGTP TriLink BioTechnologies LLC 3’-O-methyl-dCTP TriLink BioTechnologies LLC Example 2: In Silico Modeling
T331 T331M, T331M + T331M + T331M + T331M + T331M + T331S E180K, R454T, E180K + R461V, N474R, T331A, T331S + T331S + R454T, T331S + T331S + T331V, E180K, R454T, T331S + R461V, N474R, T331G, T331A + T331A + E180K + T331A + T331A + T331I, E180K, R454T, R454T, R461V, N474R, T331N, T331V + T331V + T331A + T331V + T331V + T331C, E180K, R454T, E180K + R461V, N474R, T331L T331G + T331G + R454T, T331G + T331G + E180K, R454T, T331V + R461V, N474R, T331I + T331I + E180K + T331I + T331I + E180K, R454T, R454T, R461V, N474R, T331N + T331N + T331G + T331N + T331N + E180K, R454T, E180K + R461V, N474R, T331C + T331C + R454T, T331C + T331C + E180K, R454T, T331I + R461V, N474R, T331L + T331L + E180K + T331L + T331L + E180K R454T R454T, R461V N474R T331N + E180K + R454T, T331C + E180K + R454T, T331L + E180K + R454T G332 G332A G332A + E180K G332A + G332A + G332A + G332A + R454T E180K + R461V N474R R454T G333 G333S, G333S + E180K, G333S + G333S + G333S + G333S + G333A, G333A + R454T, E180K + R461V, N474R, G333D, E180K + G333A + R454T, G333A + G333A + G333P, G333D + R454T, G333A + R461V, N474R, G333E E180K, G333D + E180K + G333D + G333D + G333P + R454T, R454T, R461V, N474R, E180K, G333P + G333D + G333P + G333P + G333E + E180K R454T, E180K + N461V, N474R, G333E + R454T, G333E + G333E + R454T G333P + N461V N474R E180K + R454T, G333E + E180K + R454T G333 and G333S + G333S + F334Y + G333S + G333S + G333S + G333S + F334 F334Y E180K F334Y + F334Y + F334Y + F334Y + R454T E180K + R461V N474R R454T F334 F334H, F334H + E180K, F334H + F334H + F334H + F334H + F334Y, F334Y + E180K, R454T, E180K + R461V, N464R, F334N F334N + E180K F334Y + R454T, F334Y + F334Y + R454T, F334Y + R461V, N474R, F334N + E180K + F334N + F334N + R454T R454T, R461V N474R F334N + E180K + R454T F334 and F334S + F334S + F334S + F334S + F334S + F334S + Y 334_335insY 334_335insY + 334_335insY + 334_335insY + 334_335insY + 334_335insY + insertion E180K R454T E180K + R461V N474R between R454T F334 and R335 R335 R335L, R335L + E180K, R335L + R335L + R335L + R335L + R335S, R335S + E180K, R454T, E180K + R461V, N474R, R335K, R335K + E180K, R335S + R454T, R335S + R335S + R335W, R335W + R454T, R335S + R461V, N474R, R335T E180K, R335K + E180K + R335K + R335K + R335P + E180K R454T, R454T, R461V, N474R, R335W + R335K + R335W + R335W + R454T, E180K + R461V, N474R, R335T + R454T, R335T + R335T + R454T R454T, R461V N474R R335W + E180K + R454T, R335T + E180K + R454T R336 R336K, R336K + E180K, R336K + R336K + R336K + R336K + R336S, R336S + E180K, R454T, E180K + R461V, N474R, R336I, R336I + E180K, R336S + R454T, R336S + R336S + R336N, R336N + R454T, R336S + R461V, N474R, R336V, E180K, R336I + E180K + R336I + R336I + R336Q R336V + E180K, R454T, R454T, R461V, N474R, R336Q + E180K R336N + R336I + R336N + R336N + R454T, E180K + R461V, N474R, R336V + R454T, R336V + R336V + R454T, R454N + R461V, N474R, R336Q + E180K + R336Q + R336Q + R454T R454T, R461V N474R R336V + E180K + R454T, R336Q + E180K + R454T G337 G337K, G337K + R337K + R336K + G337K + G337K + G337E, E180K + R454T, E180K + R461V, N474R, G337A, G337E + E180K, R337E + R454T, G337E + G337E + G337D, G337A + E180K, R454T, R336S + R461V, N474R, G337H, G337D + G337A + E180K + G337A + G337A + G337S E180K, R454T, R454T, R461V, N474R, G337H + E180K, G337D + R336I + G337D + G337D + G337H + R454T, E180K + R461V, N474R, E180K, G337H + R454T, G337H + G337H + G337S + E180K R454T, R454N, R461V, N474R, G337H + E180K + G337H + G337H + R454T, R454T, R461V, N474R, G337S + R336I + G337S + G337S + R454T E180K + R461V N474R R454T, R336V + E180K + R454T, R336Q + E180K + R454T K338 K338R, K338R + E180K, K338R + K338R + K338R + K338R + K338A K338A + E180K R454T, E180K + R461V, N474R, K338A + R454T, K338A + K338A + R454T K338A + R461V N474R E180K + R454T G341 G341C, G341C + G341C + G341C + G341C + G341C + G341S, E180K, R454T, E180K + R461V, N474R, G341V, G341S + E180K, G341S + R454T, G341S + G341S + G341I G341V + R454T, G341S + R461V, N464R, E180K, G341V + E180K + G341V + G341V + G341I + E180K R454T, R454T, R461V, N474R, G341I + G341V + G341I + G341I + R454T E180K + R461V N474R R454T, G341I + E180K + R454T H342 H342G, H342G + H342G + H342G + H342G + H342G + H342K, E180K, R454T, E180K + R461V, N474R, H342R, H342K + H342K + R454T, H342K + H342K + H342D E180K, R454T, H342K + R461V, N474R, H342R + H342R + E180K + H342R + H342R + E180K, R454T, R454T, R461V, N474R, H342D + E180K H342D + H342R + H342D + H342D + R454T E180K + R461V N474R R454T, H342D + E180K + R454T Example 3: Incorporation of dNTPs with Phosphate Blocking Groups
N304K E457R N304R D473K N509K D473R N509R Q402K D434K Q402R D434R D399K D170K D339R D170R E382K D173K E382R D173R Q455K E457K Q455R INCORPORATION BY REFERENCE
EQUIVALENTS