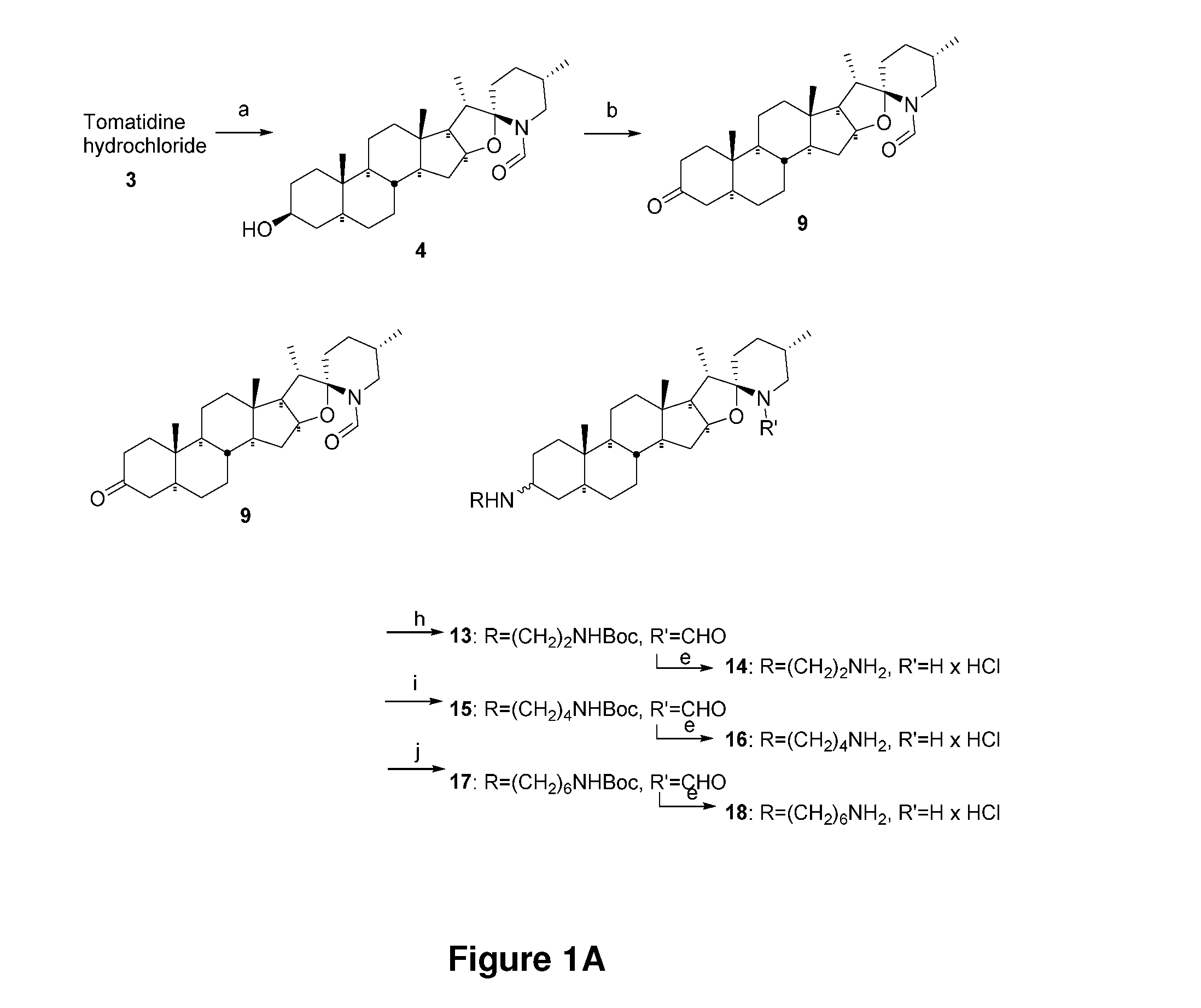
ATP SYNTHASE INHIBITORS AND STEROID ALKALOIDS AND USES THEREOF AS ANTIMICROBIAL AGENTS AND AS POTENTIATORS FOR AMINOGLYCOSIDES AGAINST PATHOGENIC BACTERIA
This application is a PCT application Serial No PCT/CA2014/0* filed on Dec. 23, 2014 and published in English under PCT Article 21(2), which itself claims benefit of U.S. provisional application Ser. No. 61/919,896, filed on Dec. 23, 2013. All documents above are incorporated herein in their entirety by reference. The present invention relates to novel antimicrobial compounds and potentiators for antimicrobial compounds. The present invention is also concerned with the use of bacterial ATP synthase inhibitors such as steroid alkaloids, as antimicrobial agents, and potentiators of the antimicrobials activity of aminoglycosides against pathogenic bacterial strains, methods of manufacturing same, disinfection, sterilization or antisepsis methods using the same. Pursuant to 37 C.F.R. 1.821(c), a sequence listing is submitted herewith as an ASCII compliant text file named 765-PCT-SEQUENCE LISTING 14692-42_ST25, that was created on Dec. 18, 2014 and having a size of 27 kilobytes. The content of the aforementioned file is hereby incorporated by reference in its entirety. Staphylococci are widely disseminated Gram-positive opportunistic bacterial pathogens responsible for many medical problems in humans, including skin and soft-tissue infections, surgical infections, endocarditis and hospital-acquired bacteriemia (Casey et al., 2007; Kloos and Bannerman, 1994). These bacteria are also the cause of several diseases in animals such as birds, cows, dogs, poultries, rabbits and others (Jacques et al., 2010; Pyorala and Taponen, 2009; Stepan et al., 2004). Staphylococci are divided in coagulase-positive species, Bacterial small-colony variants (SCVs) are derived from normal bacterial strains and show a slow-growth phenotype (i.e., they produce small colonies when cultivated on solid media). The SCV phenotype is widespread among microbes. SCVs have been described for several bacterial species and have been recovered from many different clinical specimens such as abscesses, blood, bones and joints, the respiratory tract and soft tissues (Proctor et al., 2006). For examples, SCVs were detected among the staphylococci such as Although cystic fibrosis (CF) is fundamentally a genetic disorder, the majority of patients afflicted by this disease will ultimately succumb from respiratory failure subsequent to chronic bacterial infections (Lyczak et al., 2002). More recent investigations reveal that the CF airways are colonized by complex polymicrobial communities constituted of numerous microorganisms, encompassing more bacterial species than originally thought, and suggest that interactions between these microorganisms influence the course of the disease (Sibley and Surette, 2011). Some focus has been directed toward understanding the outcome of the interactions between Gram positive bacteria, mainly staphylococci and MRSA, as well as Bovine mastitis is the most frequently occurring and costly disease affecting dairy producers. The transmittable bacterium Infections caused by antibiotic-resistant bacteria represent an overwhelming growing problem both in human and veterinary medicine. One reason explaining this widespread of drug resistances is that the currently available antibiotics have been largely designed on a limited number of chemical scaffolds, which allowed pathogens to adapt and circumvent common antibiotic action mechanisms (Shah, 2005; Talbot et al., 2006). A number of bacterial species such as It would be highly desirable to identify antibiotic compounds targeting electron transport-deficient microbes (e.g., SCVs) and/or potentiating the growth inhibitory activity of aminoglycosides against pathogenic bacteria (e.g., antibiotic-resistant bacteria and/or those causing chronic and severe infections) and/or reducing bacterial resistance development toward aminoglycosides. It would also be highly desirable to identify antibiotic compounds that can be used to reduce bacterial colonization in food, preserve food or treat infections caused by foodborne pathogens. The present description refers to a number of documents, the content of which is herein incorporated by reference in their entirety. The present invention relates in part to the discovery that specific steroid alkaloids possess not only the ability to inhibit electron-transport deficient (e.g., SCVs, bacteria affected by another organism producing inhibitors of the electron transport chain) bacteria (e.g., Firmicutes (e.g., preferably the Bacillales The present invention also shows that bacterial ATP synthase inhibitors targeting specific regions of the ATP synthase subunit C (e.g., steroid alkaloids) may be used to potentiate the antimicrobial activity of aminoglycosides (e.g., tobramycin, streptomycin, kanamycin, gentamicin, plazomycin and amikacin) against Firmicutes (e.g., preferably the Bacillales More particularly, the present invention provides in an embodiment, a compound of formula (I): wherein: In another aspect, the present invention provides a compound of formula (I): wherein: In a specific embodiment of the compound, R3 is —C(CH3)(NHR9), and R4 is H, with the proviso that when R1 is OH and R2 is H, R9 is substituted or unsubstituted alkyl. In another specific embodiment of the compound, R9 is substituted or unsubstituted alkyl. In another specific embodiment of the compound, R9 is —(CH2)qCHR15R16, wherein q is 1 to 6. In another specific embodiment of the compound, R15 and R16 are identical or different and are selected from the group consisting of H, substituted or unsubstituted alkyl, substituted or unsubstituted aryl, substituted or unsubstituted cycloalkyl, and substituted or unsubstituted aralkyl, wherein substituted alkyl, substituted aryl, substituted cycloalkyl, substituted aralkyl and substituted —(CH2)pNR13R14 comprises one or more identical or different substitutions selected from alkyl, aryl, cycloalkyl and aralkyl. In another specific embodiment of the compound, R15 and R16 are identical or different and are selected from the group consisting of H, substituted or unsubstituted alkyl. In another specific embodiment of the compound, R15 and R16 are identical or different and are selected from the group consisting of H and unsubstituted alkyl. In another specific embodiment of the compound, R15 and R16 are identical or different and are selected from the group consisting of H and —CH3. In another specific embodiment of the compound, R15 and R16 are H. In another specific embodiment of the compound, R15 and R16 are —CH3. In another specific embodiment of the compound, q is 2 or 3. In another specific embodiment of the compound, wherein q is 2. In another specific embodiment of the compound, R1 is H and R2 is —OH; or R1 is —OH and R2 is H. In another specific embodiment, the compound is (i) or (ii) or a salt, stereoisomer or any mixture of stereoisomers of (i) or (ii). In another specific embodiment, R3 and R4 form together a spirocyclic structure of formula II. In another specific embodiment, R10 is H, or substituted or unsubstituted alkyl. In another specific embodiment, R10 is H, or substituted or unsubstituted alkyl C1-C5. In another specific embodiment, R10 is H. In another specific embodiment, R1 is H and R2 is —NR5R6; or R1 is —NR5R6 and R2 is H. In another specific embodiment, R5 is H and R6 is —(CH2)nNR7R8 or R6 is H and R5 is —(CH2)nNR7R8. In another specific embodiment, R7 and R8 are identical or different and are selected from the group consisting of H and substituted or unsubstituted alkyl. In another specific embodiment, R7 and R8 are identical or different and are selected from the group consisting of H and substituted or unsubstituted alkyl C1 to C3. In another specific embodiment, R7 and R8 are H. In another specific embodiment, n is 2 to 6. In another specific embodiment, n is 2. In another specific embodiment, n is 4. In another specific embodiment, n is 6. In another specific embodiment, the compound is i) or
or a salt, stereoisomer or any mixture of stereoisomers of any one of (i) to (iii). In accordance with another aspect, the present invention provides a compound inhibiting a bacterial ATP synthase by targeting (1) a first conserved region of the bacterial ATP synthase subunit C polypeptide (“first conserved region”) defined by (A) LX1X2X3AAAIAX4GLX5ALGAGIGNGLIVX6X7TX8EGX9ARQPEX10(SEQ ID NO: 24) wherein X1, X2, X3, X4, X5, X6, X7, X8, X9, or X10 are any amino acid; or X1 is an aliphatic amino acid or absent, preferably glycine or absent; X2 is an aliphatic amino acid or absent, preferably valine or absent; X3 is an aliphatic amino acid, preferably isoleucine or leucine; X4 is an aliphatic amino acid, preferably valine or isoleucine; X5 is any amino acid, preferably an aliphatic amino acid or an hydroxyl or sulfur containing amino acid, more preferably glycine, serine or alanine; X6 is any amino acid, preferably an aliphatic amino acid or an hydroxyl or sulfur containing amino acid, preferably serine or glycine; X7 is a basic amino acid, preferably lysine or arginine; X8 is an aliphatic amino acid, preferably valine or isoleucine; X9 is an aliphatic amino acid, preferably valine or isoleucine; and X10 is an aliphatic amino acid, preferably alanine or leucine; or (B) defined by LX1X2IAAAIAX3GLX4ALGAGIGNGLIVSX5TX6EGX7ARQPEX8(SEQ ID NO: 27), wherein X1 is an aliphatic amino acid or absent, preferably glycine or absent; X2 is an aliphatic amino acid or absent, preferably valine or absent; X3 is an aliphatic amino acid, preferably valine or isoleucine; X4 is any amino acid, preferably an aliphatic amino acid or an hydroxyl or sulfur containing amino acid, more preferably glycine or serine; X5 is a basic amino acid, preferably lysine or arginine; X6 is an aliphatic amino acid, preferably valine or isoleucine; X7 is an aliphatic amino acid, preferably valine or isoleucine; and X8 is an aliphatic amino acid, preferably alanine or leucine; and/or (2) a second conserved region of the bacterial ATP synthase subunit C polypeptide (“second conserved region”), defined by GZ1Z2LVEALPIIZ3VVIAF (SEQ ID NO: 30), wherein Z1, Z2 and Z3 are any amino acid, orwherein Z1 is an aliphatic amino acid, preferably valine or isoleucine; Z2 is an aliphatic amino acid, preferably glycine or alanine; and Z3 is an aliphatic amino acid, preferably glycine or alanine, (A) in combination with an aminoglycoside antibiotic for preventing or treating a infection by a Firmicutes phylum bacteria of the order Bacillales in a subject; or (B) (a) for preventing or treating a bacterial infection caused by an electron transport-deficient bacteria in a subject; or (b) for the disinfection, sterilization and/or antisepsis of an object contaminated with an electron transport-deficient bacteria, wherein the inhibitor is not tomatidine, tomatidine 3-sulfate, tomatidine 3-phosphate, 3α-hydroxytomatidine, 3-oxo-tomatidine, 3-aminotomatidine, N-formyl tomatidine, N-formyl-3α-acetyltomatidine, 3α-hydroxytomatidine, N-formyl-3-oxotomatidine, 3-oxotomatidine, O-allyl-N-formyltomatidine, O-allyltomatidine, tomatidine methanesulfonate, tomatidine citrate, O-acetyl-N-benzylpregn-5,6-en-3β-ol-20-amine, pregnan-3β-ol-20-amine, O-acetylpregn-5,6-en-3β-ol-20-((N,N-dimethylamino)propyl)amine, pregnan-3β-ol-20-((N,N-dimethylamino)propyl)amine, O-acetylpregnan-3β-ol-20-(boc-aminoethyl)amine, O-acetylpregnan-3β-ol-20-(boc-aminopropyl)amine, O-acetylpregnan-3β-ol-20-(boc-aminobutyl)amine, pregnan-3β-ol-20-(boc-aminoethyl)amine, pregnan-3β-ol-20-(boc-aminopropyl)amine, pregnan-3β-ol-20-(boc-aminobutyl)amine, pregnan-3β-ol-20-(aminoethyl)amine, pregnan-3β-ol-20-(aminopropyl)amine, pregnan-3β-ol-20-(aminobutyl)amine, O-t-butyldimethylsilylpregnanolone, t-Butyldimethysilylpregnane-3,20-diol, pregnane-3,20-diol, pregnanolone, O-t-butyldimehylsilyl-21-bromopregnanolone, N,N-dimethyl-21-aminopregnanolone, 21-piperidinopregnanolone, N-methyl-21-aminopregnanolone, 21-piperazinopregnanolone, aminothiazole, 3β-hydroxy-20-(pyrid-2-yl)-5α-pregnane, 3β-hydroxy-20-(thiazol-2-yl)-5α-pregnane, 3β-hydroxy-20-(piperidin-2-yl)-5α-pregnane, 3β-hydroxy-20-(thiazol-4-yl)-5α-pregnane, 3β-hydroxy-20-(imidazol-4-yl)-5α-pregnane, 3-methoxymethyltomatidine, 26-amino-3β-hydroxy-16β-methoxy-21-methyl-5α-cholestano-22,26-piperidine, (25S)-26-Acetylamino-3β,16β-dihydroxy-5α-cholestane, 3β-hydroxy-20-(2-aminothiazol-4-yl)-5α-pregnane hydrochloride or a salt thereof. In accordance with another aspect, the present invention provides a compound inhibiting a bacterial ATP synthase by targeting the ATP synthase subunit C conserved region: (i) LX1X2X3AAAIAX4GLX5ALGAGIGNGLIVX6X7TX8EGX8ARQPEX10as set forth in SEQ ID NO: 24, 25 or 26; or (ii) GZ1Z2LVEALPIIZ3VVIAF as set forth in SEQ ID NO: 30, 31 or 32, (A) in combination with an aminoglycoside antibiotic for preventing or treating a infection by a Firmicutes phylum bacteria of the order Bacillales in a subject; or (B) (a) for preventing or treating a bacterial infection caused by an electron transport-deficient bacteria in a subject; or (b) for the disinfection, sterilization and/or antisepsis of an object contaminated with an electron transport-deficient bacteria, wherein the inhibitor is not tomatidine, tomatidine 3-sulfate, tomatidine 3-phosphate, 3α-hydroxytomatidine, 3-oxo-tomatidine, 3-aminotomatidine, N-formyl tomatidine, N-formyl-3α-acetyltomatidine, 3α-hydroxytomatidine, N-formyl-3-oxotomatidine, 3-oxotomatidine, O-allyl-N-formyltomatidine, O-allyltomatidine, tomatidine methanesulfonate, tomatidine citrate, O-acetyl-N-benzylpregn-5,6-en-3β-ol-20-amine, pregnan-3β-ol-20-amine, O-acetylpregn-5,6-en-3β-ol-20-((N,N-dimethylamino)propyl)amine, pregnan-3β-ol-20-((N,N-dimethylamino)propyl)amine, O-acetylpregnan-3β-ol-20-(boc-aminoethyl)amine, O-acetylpregnan-3β-ol-20-(boc-aminopropyl)amine, O-acetylpregnan-3β-ol-20-(boc-aminobutyl)amine, pregnan-3β-ol-20-(boc-aminoethyl)amine, pregnan-3β-ol-20-(boc-aminopropyl)amine, pregnan-3β-ol-20-(boc-aminobutyl)amine, pregnan-3β-ol-20-(aminoethyl)amine, pregnan-3β-ol-20-(aminopropyl)amine, pregnan-3β-ol-20-(aminobutyl)amine, O-t-butyldimethylsilylpregnanolone, t-Butyldimethysilylpregnane-3,20-diol, pregnane-3,20-diol, pregnanolone, O-t-butyldimehylsilyl-21-bromopregnanolone, N,N-dimethyl-21-aminopregnanolone, 21-piperidinopregnanolone, N-methyl-21-aminopregnanolone, 21-piperazinopregnanolone, aminothiazole, 3β-hydroxy-20-(pyrid-2-yl)-5α-pregnane, 3β-hydroxy-20-(thiazol-2-yl)-5α-pregnane, 3-hydroxy-20-(piperidin-2-yl)-5α-pregnane, 3β-hydroxy-20-(thiazol-4-yl)-5α-pregnane, 3β-hydroxy-20-(imidazol-4-yl)-5α-pregnane, 3-methoxymethyltomatidine, 26-amino-3β-hydroxy-16β-methoxy-21-methyl-5α-cholestano-22,26-piperidine, (25S)-26-Acetylamino-3β,16β-dihydroxy-5α-cholestane, 3β-hydroxy-20-(2-aminothiazol-4-yl)-5α-pregnane hydrochloride or a salt thereof. In a specific embodiment, the compound is (i) the compound, stereoisomer, mixture of stereoisomers or salt as defined above; or (ii) (R)-3-((R)-(6-bromo-2-methoxyquinolin-3-yl)(phenyl)methyl)-7-(4-methylpiperazin-1-yl)-1-phenylheptan-3-ol; (1R,2S)-1-(6-bromo-2-methoxyquinolin-3-yl)-2-(2,5-difluorophenyl)-6-(dimethylamino)-1-phenylhexan-2-ol; (R)-3-((R)-(6-bromo-2-methoxyquinolin-3-yl)(4-methoxyphenyl)methyl)-1-(dimethylamino)-5-phenylpentan-3-ol; or (S)-3-((R)-(6-bromo-2-methoxyquinolin-3-yl)(phenyl)methyl)-7-(methylamino)-1-phenylheptan-3-ol. In a specific embodiment, the compound is for preventing or treating a bacterial infection in a subject; or (b) for the disinfection, sterilization and/or antisepsis of an object contaminated by the bacteria. In another specific embodiment, the bacterial infection or contamination is caused by normal bacteria. In another specific embodiment, the compound is in combination with an aminoglycoside antibiotic for preventing or treating a bacterial infection in a subject. In another specific embodiment, the compound further comprises the use of a beta-lactam. In another specific embodiment, the bacterial infection is caused by electron-transport deficient bacteria. In another specific embodiment, the bacteria are small-colony variants (SCVs). In another specific embodiment, the bacterial infection is caused by multidrug resistant bacteria. In another specific embodiment, the bacteria are Firmicutes phylum bacteria. In another specific embodiment, the Firmicutes phylum bacteria are Bacillales. In another specific embodiment, the bacteria are of the genus In accordance with another aspect, the present invention provides a compound as defined above, or a stereoisomer, any mixture of stereoisomers or a salt thereof. In accordance with another aspect, there is provided a compound of the invention (e.g., listed in Table 1 below) or a salt, stereoisomer or any mixture of stereoisomers of such compound. In accordance with another embodiment, there is provided a compound of the invention (e.g., listed in Table 1 below) which has a moderate to high potentiation activity and or a moderate to high antibacterial activity or a salt, stereoisomer or any mixture of stereoisomers of such compound. Compositions and kits In accordance with another aspect, there is provided a composition comprising at least one of the compounds as defined herein, and (i) an antibiotic; (ii) an antiseptic; (iii) a disinfectant; (iv) a diluent; (v) an excipient; (vi) a pharmaceutically acceptable carrier; or (vii) any combination of (i)-(vi). In a specific embodiment, said composition is a pharmaceutical composition. In another specific embodiment, the composition comprises (i) the compound as defined herein; and (ii) an antibiotic, which is an aminoglycoside antibiotic. In another specific embodiment, the composition further comprises a beta-lactam antibiotic. In accordance with another aspect of the present invention, there is provided a kit comprising the compound defined herein or the above-mentioned composition, and instructions to use same in the prevention or treatment of a bacterial infection. In a specific embodiment of the kit, the kit comprises: (i) one or more compounds defined herein; and/or (ii) one or more compositions defined herein, and instructions to use same in the prevention or treatment of a microbial infection. In another specific embodiment of the kit, the kit further comprises (iii) an antiseptic; (iv) a disinfectant; (v) a diluent; (vi) an excipient; (vii) a pharmaceutically acceptable carrier; or (viii) any combination of (iii)-(vii). In another specific embodiment of the kit, the kit comprises: (a) an antibiotic; (b) an antiseptic; (c) a disinfectant; (d) any combination of (a)-(c). In accordance with another aspect, there is provided a kit comprising the compound as defined herein, and instructions to use same in (a) the prevention or treatment of a bacterial infection; or (b) the disinfection, sterilization and/or antisepsis of an object. In a specific embodiment, the kit further comprises an aminoglycoside antibiotic. In another specific embodiment, the kit further comprises a beta-lactam antibiotic. In another specific embodiment, the kit further comprises: (iii) an antiseptic; (iv) a disinfectant; (v) a diluent; (vi) an excipient; (vii) a pharmaceutically acceptable carrier; or (viii) any combination of (iii)-(vii). In another specific embodiment, the kit further comprises: (i) an antibiotic; (ii) an antiseptic; (iii) a disinfectant; or (iv) any combination of (i)-(iii). More specifically, in accordance with another aspect of the present invention, there is provided a kit comprising the compound as defined above, and instructions to use same in (a) the prevention or treatment of a microbial infection; or (b) the disinfection, sterilization and/or antisepsis of an object. In a specific embodiment of the kit, the kit further comprises an aminoglycoside antimicrobial agent. In another specific embodiment of the kit, the aminoglycoside antimicrobial agent is amikacin, gentamicin, kanamycin, streptomycin, tobramycin or plazomycin. In another specific embodiment of the kit, the kit further comprises a beta-lactam antimicrobial agent. In a specific embodiment of the composition or kit, the aminoglycoside antimicrobial agent is amikacin, gentamicin, kanamycin, streptomycin, tobramycin or plazomycin. In a specific embodiment of the composition, the composition further comprises a beta-lactam antimicrobial agent. In a specific embodiment of the composition, the composition comprises a compound as defined herein or a salt, stereoisomer or any mixture of stereoisomers of such compound. In another specific embodiment of the composition, the composition comprises a compound as listed in Table 1 below or a salt, stereoisomer or any mixture of stereoisomers of such compound. In accordance with yet another embodiment, the composition comprises a compound as listed in Table 1 below which has a moderate to high potentiation activity and or a moderate to high antibacterial activity or a salt, stereoisomer or any mixture of stereoisomers of such compound. In another specific embodiment of the compounds for uses (or corresponding methods or uses), said subject or object is food, a cow or a human. In another specific embodiment of the compounds for uses (or corresponding methods or uses), said subject is a human. In accordance with another aspect of the present invention, there is provided a method of identifying a pathogen, the microbial infection of which is treatable by the compound as defined herein or a composition comprising the compound, said method comprising contacting said bacterial pathogen with said compound or composition and determining the effect of said compound or composition on the growth or survival of said pathogen, wherein a decrease in the growth or survival of said pathogen in the presence as compared to in the absence of said compound or composition is an indication that said bacterial pathogen is treatable by said compound or composition. In a specific embodiment of the method, use and compositions for uses of the present invention, said subject is an animal (e.g., cattle such as cow; goat, ewe, ass, horse, pig, cat, dog, etc.). In another specific embodiment, said subject is a cow. In another specific embodiment, said subject is a human. Other advantages and features of the present invention will become more apparent upon reading of the following non-restrictive description of specific embodiments thereof, given by way of example only with reference to the accompanying drawings. In the appended drawings: In certain embodiments, the present invention relates to the unexpected discovery that inhibitors of bacterial ATP synthase targeting specific regions of the subunit C of the enzyme have a very potent growth inhibitory activity against not only electron transport-deficient bacteria but also against normal (i.e. non electron-transport deficient) bacteria (e.g., firmicutes phylum (e.g., preferably the Bacillales The present invention also encompasses using a compound of the present invention with at least another active ingredient (e.g., another antibiotic agent). Compounds of the present invention may be used as antimicrobial compounds against microbial targets described herein, through their ATP synthase inhibitory activity or through other mechanisms. In other embodiments, the present invention also relates to the surprising discovery that bacterial ATP synthase inhibitors targeting specific regions of subunit C of the ATP synthase enzyme selectively potentiate the inhibitory activity of aminoglycoside antimicrobial agents against normal (i.e. non electron transport-deficient (e.g., non-SCVs)) bacteria (e.g., firmicutes (e.g., preferably the Bacillales Compounds of the present invention may be used as potentiators of the inhibitory activity of aminoglycoside antimicrobial agents against microbial targets described herein, through their ATP synthase inhibitory activity or through other mechanisms. The use of the word “a” or “an” when used in conjunction with the term “comprising” in the claims and/or the specification may mean “one” but it is also consistent with the meaning of “one or more”, “at least one”, and “one or more than one”. As used herein the term “microbe” includes without being limited to a bacterium. As used here in the term “infection” refers to a monomicrobic or a polymicrobic infection. It refers to infections involving at least one microbial target of the present invention (e.g., a normal bacteria (e.g., firmicutes (e.g., preferably the Bacillales As used herein the terms “polymicrobic infection” are interchangable with the terms “mixed infection”, “co-infection” or “polymicrobial infection”. As used herein, they refer to a co-culture, an infection, a colonization, a community or a population of microbes of different species or strains found together either as planktonic organisms or embedded in a biofilm structure. More particularly, polymicrobic infections targeted by compounds of the present invention include at least one microorganism (e.g., bacteria) producing at least one electron transport inhibitor (e.g., The use of the word “bacterium” in this specification and claim(s) may be interchanged with the words “bacteria”, “bacterial pathogen”, “infectious agent”, “strain” or “bacterial strain” (e.g., living either as planktonic microorganism, embedded in a biofilm structure or intracellular). As used herein the terms “reducing the development of resistance” toward an antimicrobial agent (e.g., aminoglycoside) refers to a reduction in the number of bacteria that become resistant to the antimicrobial agent when treated with the antimicrobial agent in combination with a compound of the present invention as compared to when treated with the antimicrobial agent alone. As used herein the term “reduce”, “reduction” or “decrease” or “prevention” of development of resistance toward an antimicrobial agent refers to a reduction in development of resistance toward an antimicrobial agent of at least 10% as compared to reference (e.g., treatment with antimicrobial agent alone) development of resistance, in an embodiment of at least 20% lower, in a further embodiment of at least 30%, in a further embodiment of at least 40%, in a further embodiment of at least 50%, in a further embodiment of at least 60%, in a further embodiment of at least 70%, in a further embodiment of at least 80%, in a further embodiment of at least 90%, in a further embodiment of 100% (complete prevention). As used herein, the words “comprising” (and any form of comprising, such as “comprise” and “comprises”), “having” (and any form of having, such as “have” and “has”), “including” (and any form of including, such as “includes” and “include”) or “containing” (and any form of containing, such as “contains” and “contain”) are inclusive or open-ended and do not exclude additional, un-recited elements or method steps. As used herein, the terms “molecule”, “compound” and “agent” are used interchangeably and broadly to refer to natural, synthetic or semi-synthetic molecules or compounds. The term “compound” therefore denotes, for example, chemicals, macromolecules, cell or tissue extracts (from plants or animals) and the like. Non-limiting examples of compounds include peptides, antibodies, carbohydrates, nucleic acid molecules and pharmaceutical agents. The compound can be selected and screened by a variety of means including random screening, rational selection and by rational design using, for example, ligand modeling methods such as computer modeling. As will be understood by the person of ordinary skill, molecules having non-naturally occurring modifications are also within the scope of the term “compound”. For example, the compounds of the present invention can be modified to enhance their activity, stability, and/or bioavailability, and also to lower its toxicity. The compounds or molecules identified in accordance with the teachings of the present invention have a therapeutic value in diseases or conditions related to microbial infections. As used herein, the terms “bacterial ATP synthase subunit C” refers to an ATP synthase subunit C defined by the sequence as set forth in SEQ ID NO: 22 (consensus derived from ATP synthase subunit C polypeptides of bacterial species depicted in In another embodiment, each X refers to any amino acid in the corresponding position of the ATP synthase subunit C polypeptide sequence of As used herein, the terms “bacterial ATP synthase inhibitor targeting the ATP synthase subunit C” refer to any inhibitor that specifically targets or interacts with (1) a first conserved region of the bacterial ATP synthase subunit C polypeptide (“first conserved region”) defined by (A) LX1X2X3AAAIAX4GLX5ALGAGIGNGLIVX6X7TX8EGX8ARQPEX10(SEQ ID NO: 24) wherein X1, X2, X3, X4, X5, X6, X7, X8, X9, or X10 are any amino acid; or X1 is an aliphatic amino acid or absent, preferably glycine or absent; X2 is an aliphatic amino acid or absent, preferably valine or absent; X3 is an aliphatic amino acid, preferably isoleucine or leucine; X4 is an aliphatic amino acid, preferably valine or isoleucine; X5 is any amino acid, preferably an aliphatic amino acid or an hydroxyl or sulfur containing amino acid, more preferably glycine, serine or alanine; X6 is any amino acid, preferably an aliphatic amino acid or an hydroxyl or sulfur containing amino acid, preferably serine or glycine; X7 is a basic amino acid, preferably lysine or arginine; X8 is an aliphatic amino acid, preferably valine or isoleucine; X9 is an aliphatic amino acid, preferably valine or isoleucine; and X10 is an aliphatic amino acid, preferably alanine or leucine; or (B) defined by LX1X2IAAAIAX3GLX4ALGAGIGNGLIVSX5TX6EGX7ARQPEX8(SEQ ID NO: 27), wherein X1 is an aliphatic amino acid or absent, preferably glycine or absent; X2 is an aliphatic amino acid or absent, preferably valine or absent; X3 is an aliphatic amino acid, preferably valine or isoleucine; X4 is any amino acid, preferably an aliphatic amino acid or an hydroxyl or sulfur containing amino acid, more preferably glycine or serine; X5 is a basic amino acid, preferably lysine or arginine; X6 is an aliphatic amino acid, preferably valine or isoleucine; X7 is an aliphatic amino acid, preferably valine or isoleucine; and X8 is an aliphatic amino acid, preferably alanine or leucine. Preferably within this region, it targets any subregion of 37, 36, 35, 34, 33, 32, 31, 30, 29, 28, 27, 26, 25, 24, 23, 22, 21, 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, 1 amino acid residue(s). More preferably, within the first conserved region, the inhibitor targets the subregion ALGAGIG (SEQ ID NO: 33); even more preferably within this region, it targets the subregion LGAGI (SEQ ID NO: 34), ALGAG (SEQ ID NO: 35) or GAGIG (SEQ ID NO: 36); more preferably within this region, it targets the subregion LGA, GAG or AGI; even more preferably within this region, it targets the subregion GA or AG; and even more preferably, it targets the A residue (underlined in the full conserved region). For instance in the strains In a specific embodiment, the ATP synthase inhibitors of the present invention specifically target bacterial ATP synthase (and not, or to a lesser extent, other types of ATP synthases such as e.g., mitochondrial ATP synthases). Exemplified compounds of the present invention represent some examples of such ATP synthase inhibitors. As used herein the term “aryl” unless otherwise expressly characterized, may refer to a substituted or unsubstituted aryl (e.g., C5-C6, C5 or C6) (which may be an heterocycle i.e. one or more carbon replaced by another atom (e.g., N, O, S, etc.), wherein the substitutent(s), if any, is one or more identical or different substituents selected from an alkyl, aryl, aralkyl, which alkyl, aryl, aralkyl may be substituted or not with one or more alkyl, aryl, cycloalkyl and/or aralkyl, halide, OH, OMe, NO2, NH2and/or CO2H, including heterocycles. Herein, the term “substituted aryl” is sometimes expressly used. In a specific embodiment, the substitutent(s) may be one or more identical or different substitutents selected from an alkyl, aryl, cycloalkyl and/or aralkyl. As used herein the term “alkyl” unless otherwise characterized, may refer to saturated or unsaturated (e.g., allyle), substituted or unsubstituted, and/or linear or branched alkyl (C1 to C10, or C2 to C9, or C3 to C8, of C4 to C7, or C5 to C6, or C1, C2, C3, C4, C5, C6, C7, C8, C9 or C10), wherein the substitutent(s), if any, is one or more identical or different substituents selected from an alkyl, aryl, aralkyl, which alkyl, aryl, aralkyl may be substituted or not with one or more alkyl, aryl and/or aralkyl, halide, OH, OMe, NO2, NH2and/or CO2H. Without being so limited, it includes —CH2—CH═CH2, and —(CH2)3—CH(CH3)CH2. Herein, the term “substituted alkyl” is sometimes also expressly used. In a specific embodiment, the substitutent(s) may be one or more identical or different substitutents selected from an alkyl, aryl, cycloalkyl and/or aralkyl. As used herein the term “aralkyl” unless otherwise expressly characterized, refers to a radical derived from an alkyl radical by replacing one or more hydrogen atoms by aryl groups. It includes saturated or unsaturated, substituted or unsubstituted, linear or branched aralkyl (C1 to C10, or C2 to C9, or C3 to C8, of C4 to C7, or C5 to C6, or C1, C2, C3, C4, C5, C6, C7, C8, C9 or C10), wherein the substitutent, if any, is one or more identical or different substituents selected from an alkyl, aryl, aralkyl, which alkyl, aryl, aralkyl may be substituted or not with one or more alkyl, aryl and/or aralkyl halide, OH, OMe, NO2, NH2or CO2H. Herein, the term “substituted aryl” is sometimes also expressly used. In a specific embodiment, the substitutent(s) may be one or more identical or different substitutents selected from an alkyl, aryl, cycloalkyl and/or aralkyl. As used herein the term “cycloalkyl” unless otherwise expressly characterized, refers to refer to a substituted or unsubstituted cycloalkyl (e.g., C3-C7, C3, C4, C5, C6, or C7) (which may be an heterocycle i.e. one or more carbon replaced by another atom (e.g., N, O, S, etc.)), wherein the substitutent(s), if any, is one or more identical or different substituents selected from an alkyl, aryl, aralkyl, which alkyl, aryl, aralkyl may be substituted or not with one or more alkyl, aryl and/or aralkyl, halide, OH, OMe, NO2, NH2and/or CO2H. Herein, the term “substituted cycloalkane” is sometimes also expressly used. In a specific embodiment, the substitutent(s) may be one or more identical or different substitutents selected from an alkyl, aryl, cycloalkyl and/or aralkyl. As used herein the terms “substituted —(CH2)nNR7R8” refer to —(CH2)nNR7R8 that may comprise one or more identical or different substitutents selected from an alkyl, aryl and/or aralkyl, which alkyl, aryl, aralkyl may be substituted or not with one or more alkyl, aryl cycloalkyl and/or aralkyl. As used herein the terms “substituted —(CH2)mNR11R12” refer to —(CH2)mNR11R12 that may comprise one or more identical or different substitutents selected from an alkyl, aryl and/or aralkyl, which alkyl, aryl, aralkyl may be substituted or not with one or more alkyl, aryl, cycloalkyl and/or aralkyl. As used herein the terms “substituted —(CH2)pNR13R14” refer to —(CH2)pNR13R14 that may comprise one or more identical or different substitutents selected from an alkyl, aryl and/or aralkyl, which alkyl, aryl, aralkyl may be substituted or not with one or more alkyl, aryl, cycloalkyl and/or aralkyl. As used herein the term <<CO>> refers to a carbonyl. The symbols used herein to denote the orientation of the hydrogen atoms are those used in the tomatidine formula presented below at the left, wherein “=” denotes □-H and “•”β-H. They are used to identify the stereochemistry of tertiary carbons (having three direct neighbors other than hydrogens). The classical representation of the hydrogens is shown in the right formula for tomatidine for comparison purposes. Such convention is used to simplify the formulas. As used herein the term “aminoglycoside” refers to an aminoglycoside antimicrobial agent and include without being so limited to amikacin, arbekacin, gentamicin, kanamycin, dideoxykanamycin, neomycin, neamine, lividomycin, butirosin, netilmicin, paromomycin, rhodostreptomycin, streptomycin, tobramycin, framycetin, ribostamycin, bekanamycin, dibekacin, hygromycin B, sisomicin, isepamicin, verdamicin, astromicin, apramycin, fortimycin, sorbistin, kasugamycin, istamycin, sagamicin, spectinomycin and other known aminoglycosides. The term aminoglycoside also includes herein the 4,5-disubstituted deoxystreptamines, 4,6-disubstituted deoxystreptamines, aminocyclitols, streptidines, actinanimes, deoxystreptamines, destomycins. It also includes neoglycosides or “next-generation aminoglycosides” (e.g., plazomycin, ACHN-490) namely aminoglycosides able to circumvent bacterial resistance mechanisms used against previous aminoglycosides. As used herein the term “combination” when used in reference to the use of the compound of the invention in combination with at least one other antibiotic (e.g., aminoglycoside) means i) simultaneously (e.g., in separate compositions or a single composition); ii) simultaneously as a single dual action compound (e.g., a conjugate of the two or more, the compound of the invention chemically linked with at least another antibiotic) in a single composition; or iii) subsequently (e.g., in separate compositions wherein the compound of the present invention is administered before (e.g., immediately before) or after (e.g., immediately after) the at least other antibiotic). The present invention encompasses therefore the use of a combination of two, three or more active ingredients including at least one compound of the present invention. A combination of three compounds in accordance of the present invention can include a compound of the present invention, an aminoglycoside and a beta-lactam (e.g., Ubrolexin™ (i.e. cefalexin [or cephalexin] and kanamycin)). Compounds of the present invention may be used as antimicrobial agents. In this respect, the compounds of the present invention are used against normal microbes (e.g., bacteria) and “electron transport-deficient microbes”. The compounds of the present invention may be used against normal or electron-transport deficient bacteria, preferably of the Firmicutes phylum. While there are currently more than 274 genera within the Firmicutes phylum, notable genera of Firmicutes include the Bacilli, order Bacillales, As used herein the term “electron transport-deficient microbes” refers for example to SCVs that have a defect in the electron transport chain, and to bacteria of a polymicrobic infection that have been affected by at least one electron transport inhibitor and/or at least one molecule related to 4-hydroxy-2-alkylquinolines produced by at least one microorganism (e.g., bacteria) (e.g., SCVs may have a defect in the electron transport chain caused by mutation, sub-optimal expression, sub-optimal biosynthesis or alteration of electron transport proteins, necessary coenzymes, cofactors or precursors, a defect in the bacterial FOF1-ATPase or proton pumps or an overall reduction of certain metabolic pathways such as the tricarboxilic cycle that ultimately affects and reduces electron transport. SCVs of a variety of bacterial species of human or animal origins are thus microbial targets of the compounds of the present invention. In a specific embodiment, the SCV is a Firmicutes phylum bacterium. The term “electron transport-deficient microbes” also refers to bacteria of a polymicrobic infection that are affected by at least one electron transport inhibitor and/or at least one molecule related to 4-hydroxy-2-alkylquinolines produced by at least one microorganism (e.g., bacteria (e.g., Compounds of the present invention may also be used as potentiators of antimicrobial agents. As used herein, the term “potentiator” in the context of an “antimicrobial agent potentiator” refers to an agent which increases the antimicrobial activity of another antimicrobial agent on a bacterium and thus creates a synergy, i.e., the activity of the combination of agents is superior to that observed for either agent individually. In this respect, the compounds of the present invention may be used in combination with aminoglycosides against “normal” (i.e. non electron transport-deficient) bacterial targets of human or animal origins that include but are not limited to Firmicutes phylum bacteria as described above (e.g., staphylococci, In a particular embodiment, the compounds of the present invention are used as potentiators of aminoglycosides against normal staphylococcal strains (e.g., As used herein the term “object” refers to an animal or to an animal tissue (e.g., skin, hands), an animal cells (e.g., in cell cultures for laboratory purpose or for use for administration to subjects), food (e.g., packaged food preparation, meat, milk, milk products, etc.), a synthetic material or a natural material. Synthetic materials include, without being so limited, working surfaces (e.g., table, counter), instruments, prosthetic devices and biomaterials. The term “Natural material” includes, without being so limited, skin grafts, tissue cultures and organs. As used herein the term “subject” or “patient” refers to an animal, preferably a mammal such as but not limited to a human, cow, goat, ewe, ass, horse, pig, chicken, cat, dog, etc. who is the object of treatment, observation or experiment. As used herein, the terms “pharmaceutically acceptable” refer to molecular entities and compositions that are physiologically tolerable and do not typically produce an allergic or similar untoward reaction, such as gastric upset, dizziness and the like, when administered to animals (e.g., cows, humans). Preferably, as used herein, the term “pharmaceutically acceptable” means approved by regulatory agency of the federal or state government or listed in the U.S. Pharmacopeia or other generally recognized pharmacopeia for use in animals, and more particularly in humans. The term “carrier” refers to a diluent, adjuvant, excipient, or vehicle with which the compounds of the present invention may be administered. Sterile water or aqueous saline solutions and aqueous dextrose and glycerol solutions may be employed as carriers, particularly for injectable solutions. Suitable pharmaceutical carriers are described in “Remington's Pharmaceutical Sciences” by E.W. Martin. Compounds of the invention may be administered in a pharmaceutical composition. Pharmaceutical compositions may be administered in unit dosage form. Any appropriate route of administration may be employed, for example, transdermal (topical), parenteral, subcutaneous, intramuscular, intramammary, intracranial, intraorbital, ophthalmic, intraventricular, intracapsular, intraarticular, intraspinal, intracisternal, intraperitoneal, intranasal, aerosol, or oral administration. Examples of specific routes of administration include parenteral, e.g., intravenous, intradermal, subcutaneous, intramammary; oral (e.g., inhalation); transdermal (topical); transmucosal, and rectal administration. Conventional pharmaceutical practice may be employed to provide suitable formulations or compositions to administer such compositions to patients. Methods well known in the art for making pharmaceutical compositions and formulations are found in, for example, Remington: The Science and Practice of Pharmacy, (20th ed.) ed. A. R. Gennaro A R., 2000, Lippincott: Philadelphia. Therapeutic formulations for oral administration, may be in the form of tablets or capsules; for transmucosal (e.g., rectal, intranasal) or transdermal/percutaneous administration may be in the form of ointments, powders, nebulized or non-nebulized dry powders, nasal drops, sprays/aerosols or suppositories; for topical administration, may be in the form of ointments, creams, gels or solutions; for parenteral administration (e.g., intravenously, intramuscularly, intradermal, intramammary, subcutaneously, intrathecally or transdermally), using for example injectable solutions. Furthermore, administration can be carried out sublingually or as ophthalmological preparations or as an aerosol, for example in the form of a spray. Intravenous, intramuscular or oral administration is generally a preferred form of use. A spray or nebulized or non-nebulized dry powders is a preferred form of use for rapid local administration in the lungs. Ointments, creams, gels or solutions added or not to bandages are preferred forms for treatment of burn wounds. The pharmaceutical compositions of the present invention may also contain excipients/carriers such as preserving agents, solubilizing agents, stabilizing agents, wetting agents, emulsifiers, sweeteners, colorants, odorants, salts for the variation of osmotic pressure, buffers, coating agents or antioxidants. As mentioned earlier, they may also contain other therapeutically valuable agents. For the purpose of oral therapeutic administration, the active compound can be incorporated with excipients and used for example in the form of tablets, troches, dragees, hard or soft gelatin capsules, solutions (e.g., syrups), aerosols, emulsions or suspensions, or capsules. For the preparation of formulations for oral administration, the compounds of the present invention may be admixed with pharmaceutically inert, inorganic or organic excipients (e.g., pharmaceutically compatible binding agents, and/or adjuvant). The tablets, pills, capsules, troches and the like can contain any of the following ingredients, or compounds of a similar nature: a binder such as microcrystalline cellulose, gum tragacanth or gelatin; an excipient such as starch or lactose, a disintegrating agent such as alginic acid, Primogel, or corn starch; a lubricant such as magnesium stearate or Sterotes; a glidant such as colloidal silicon dioxide; a sweetening agent such as sucrose or saccharin; or a flavoring agent such as peppermint, methyl salicylate, or orange flavoring. Examples of suitable excipients for tablets, dragees or hard gelatin capsules for example include lactose, maize starch or derivatives thereof, talc or stearic acid or salts thereof. Suitable excipients for use with soft gelatin capsules include for example vegetable oils, waxes, fats, semi-solid or liquid polyols etc.; according to the nature of the active ingredients it may however be the case that no excipient is needed at all for soft gelatin capsules. For the preparation of solutions and syrups, excipients which may be used include for example water, polyols, saccharose, invert sugar and glucose. For administration by inhalation through the mouth or nose, the compounds may be delivered in the form of an aerosol spray from pressured container or dispenser which contains a suitable propellant, e.g., a gas such as carbon dioxide, or a nebulizer. Nebulized or non-nebulized dry powders may be inhaled. Formulations for inhalation may contain excipients, for example, lactose, or may be aqueous solutions containing, for example, polyoxyethylene-9-lauryl ether, glycocholate and deoxycholate, or may be oily solutions for administration in the form of nasal drops, or as a gel. For transmucosal or transdermal administration, penetrants appropriate to the barrier to be permeated are used in the formulation. Such penetrants are generally known in the art, and include, for example, for transmucosal administration, detergents, bile salts, and fusidic acid derivatives. Transmucosal administration can be accomplished through the use of nasal sprays or suppositories. For transdermal administration, the active compounds are formulated into ointments, salves, gels, or creams as generally known in the art. For suppositories, and local or percutaneous application, excipients which may be used include for example natural or hardened oils, waxes, fats and semi-solid or liquid polyols. Pharmaceutical compositions suitable for injectable use include sterile aqueous solutions (where water soluble) or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersion. Solutions or suspensions used for parenteral application can include the following components: a sterile diluent such as water for injection (where water soluble), saline solution, fixed oils (e.g., paraffin oil), polyalkylene glycols such as polyethylene glycols, glycerine, propylene glycol or other synthetic solvents, oils of vegetable origin, or hydrogenated napthalenes; antibacterial agents such as benzyl alcohol or methyl parabens; antioxidants such as ascorbic acid or sodium bisulfite; chelating agents such as ethylenediaminetetraacetic acid; reducing agents such as dithiothreitol, buffers such as acetates, citrates or phosphates and agents for the adjustment of tonicity such as sodium chloride or dextrose. The pH can be adjusted with acids or bases, such as hydrochloric acid or sodium hydroxide. Biocompatible, biodegradable lactide polymer, lactide/glycolide copolymer, or polyoxyethylene-polyoxypropylene copolymers may be used to control the release of the compounds. Other potentially useful parenteral delivery systems for compounds of the invention include ethylenevinyl acetate copolymer particles, osmotic pumps, implantable infusion systems, and liposomes. The parenteral preparation can also be enclosed in ampoules, disposable syringes or multiple dose vials made of glass or plastic. For intravenous or intramammary administration, suitable carriers include physiological saline, bacteriostatic water, Cremophor EL™ (BASF, Parsippany, N.J.) or phosphate buffered saline (PBS). Liposomal suspensions (including liposomes targeted to specific cell types) can also be used as pharmaceutically acceptable carriers. A variety of liposomal formulations suitable for delivering a compound to an animal have been described and demonstrated to be effective in delivering a variety of compound, including, e.g., small molecules, nucleic acids, and polypeptides. As mentioned earlier, medicaments containing the compounds of the present invention are also an object of the present invention, as is a process for the manufacture of such medicaments, which process comprises bringing one or more of the compounds of the present invention to, if desired, one or more other therapeutically valuable substances into a galenical administration form. The compounds of the present invention may include protective groups. As used herein, and without being so limited, the term “protective group” is meant to refer to a substitutent on a heteroatom that may be cleaved in specified reaction conditions to unmask the heteroatom and includes without being so limited tert-butoxycarbonyle (BOC), t-butyldimethylsilyl (TBDMS), methoxymethyl (MOM), etc. Further examples of protecting groups may be found in Protective groups in organic synthesis, 4th edition, Peter G. M. Wuts & Theodora W. Greene editors, Wiley 2007. The compounds of the present invention include pharmacologically acceptable salts and ester derivatives thereof as well as hydrates or solvates thereof and all stereoisomeric forms of the referenced compounds. The compounds and pharmacologically acceptable esters thereof of the present invention can form pharmacologically acceptable salts if necessary. The terms “pharmacologically acceptable salt thereof” refer to a salt to which the compounds of the present invention can be converted. Preferred examples of such a salt include alkali metal salts such as a sodium salt, a potassium salt, a lithium salt, magnesium or calcium salts; alkaline earth metal salts such as a calcium salt and a magnesium salt; metal salts such as an aluminium salt, an iron salt, a zinc salt, a copper salt, a nickel salt and a cobalt salt; amine salts such as inorganic salts including an ammonium salt; organic salts or ammonium salts such as a t-octylamine salt, a dibenzylamine salt, a morpholine salt, a glucosamine salt, a phenylglycine alkyl ester salt, an ethylenediamine salt, an N-methylglucamine salt, a guanidine salt, a diethylamine salt, a triethylamine salt, a dicyclohexylamine salt, an N,N′-dibenzylethylenediamine salt, a chloroprocaine salt, a procaine salt, a diethanolamine salt, an N-benzyl-phenethylamine salt, a piperazine salt, a tetramethylammonium salt and a tris(hydroxymethyl)aminomethane salt; inorganic acid salts such as hydrohalic acid salts such as a hydrofluoride, a hydrochloride, a hydrobromide or a hydroiodide, a nitrate, a perchlorate, a sulfate or a phosphate; lower alkanesulfonates such as a methanesulfonate (mesylate), trifluoromethanesulfonate or an ethanesulfonate; arylsulfonates such as a benzenesulfonate or a p-toluenesulfonate and the like, which are non toxic to living organisms; organic acid salts such as an acetate, a malate, adipate, a fumarate, a succinate, a citrate, alginate, ascorbate, benzoate, benzenesulfonate, bisulfate, butyrate, camphorate, camphorsulfonate, cinnamate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, glucoheptanoate, glycerophosphate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, itaconate, lactate, maleate, mandelate, sulfonate, methanesulfonate, trifluoromethanesulfonates, ethanesulfonates 2-naphthalenesulfonate, nicotinate, nitrate, oxalate, pamoate, pectinate, persulfate, 3-phenylpropionate, picrate, pivalate, propionate, tartrate, thiocyanate, tosylate, and undecanoate, a tartrate, an oxalate or a maleate; and amino acid salts such as a glycine salt, a lysine salt, an arginine salt, an omithine salt, histidine, a glutamate or an aspartate salt. Additionally, basic nitrogen containing groups may be quaternized with such agents as lower alkyl halides such as methyl, ethyl, propyl, and butyl chlorides, bromides and iodides; dialkyl sulfates including dimethyl, diethyl, and dibutyl sulfate; and diamyl sulfates, long chain halides such as decyl, lauryl, myristyl and strearyl chlorides, bromides and iodides, aralkyl halides including benzyl and phenethyl bromides, and others. For further example, see S. M. Berge, et al. “Pharmaceutical Salts,” J. Pharm. Sci. 1977, 66, 1-19. Such salts can be formed quite readily by those skilled in the art using standard techniques. More specific examples of the salts formed with an acidic group present in the compounds of the present invention include metal salts such as alkali metal salts (e.g., sodium salts, potassium salts and lithium salts), alkali earth metal salts (e.g., calcium salts and magnesium salts), aluminum salts and iron salts; amine salts such as inorganic amine salts (e.g., ammonium salts) and organic amine salts (e.g., t-octylamine salts, dibenzylamine salts, morpholine salts, glucosamine salts, phenylglycinealkyl ester salts, ethylenediamine salts, N-methylglucamine salts, guanidine salts, diethylamine salts, triethylamine salts, dicyclohexylamine salts, N,N′-dibenzylethylenediamine salts, chloroprocaine salts, procaine salts, diethanolamine salts. N-benzylphenethylamine salts, piperazine salts, tetramethylammonium salts and tris(hydroxymethyl)aminomethane salts; and amino acid salts such as glycine salts, lysine salts, arginine salts, ornithine salts, glutamates and aspartates. All salts are intended to be pharmaceutically acceptable salts within the scope of the invention and all salts are considered equivalent to the free forms of the corresponding compounds for purposes of the invention. Physiologically/pharmaceutically acceptable esters are also useful as active medicaments. The term “pharmaceutically acceptable esters” embraces esters of the compounds of the present invention, in which hydroxy groups (e.g., in carboxylic acid) have been converted to the corresponding esters and may act as a prodrug which, when absorbed into the bloodstream of a warm-blooded animal, may cleave in such a manner as to release the drug form and permit the drug to afford improved therapeutic efficacy. Such esters can be formed with inorganic or organic acids such as nitric acid, sulphuric acid, phosphoric acid, citric acid, formic acid, maleic acid, acetic acid, succinic acid, tartaric acid, methanesulphonic acid, p-toluenesulphonic acid and the like, which are non toxic to living organisms. Further examples are the esters with aliphatic or aromatic acids such as acetic acid or with aliphatic alcohol (e.g., alkyl esters, including methyl, ethyl, propyl, isopropyl, butyl, isobutyl or pentyl esters, and the like) or aromatic alcohols (e.g., benzyl ester). Esters can be prepared from their corresponding acids or salts by a variety of methods known to those skilled in the art, such as, for example, by first transforming the acid to the acid chloride and then reacting the acid chloride with a suitable alcohol. Other suitable methods for making esters are described in Kemp and Vellaccio, 1980. Where esters of the invention have a basic group, such as an amino group, the compound can be converted to a salt by reacting it with an acid, and in the case where the esters have an acidic group, such as a sulfonamide group, the compound can be converted to a salt by reacting it with a base. The compounds of the present invention encompass such salts. Salts and esters of the compounds of the present invention may be prepared by known method by employing appropriate starting materials or intermediate compounds that are readily available and/or are described herein. Generally, a desired salt of a compound of this invention can be prepared in situ during the final isolation and purification of a compound by means well known in the art. For example, a desired salt can be prepared by separately reacting the purified compound in its free base or free acid form with a suitable organic or inorganic acid, or suitable organic or inorganic base, respectively, and isolating the salt thus formed. In the case of basic compounds, for example, the free base is treated with anhydrous HCl in a suitable solvent such as THF, and the salt isolated as a hydrochloride salt. In the case of acidic compounds, the salts may be obtained, for example, by treatment of the free acid with anhydrous ammonia in a suitable solvent such as ether and subsequent isolation of the ammonium salt. These methods are conventional and would be readily apparent to one skilled in the art. The compounds of this invention may be esterified by a variety of conventional procedures including reacting the appropriate anhydride, carboxylic acid or acid chloride with the alcohol group of a compound of this invention. The appropriate anhydride is reacted with the alcohol in the presence of a base to facilitate acylation such as 1,8-bis[dimethylamino]naphthalene or N,N-dimethylaminopyridine. Or, an appropriate carboxylic acid can be reacted with the alcohol in the presence of a dehydrating agent such as dicyclohexylcarbodiimide, 1-[3-dimethylaminopropyl]-3-ethylcarbodiimide or other water soluble dehydrating agents which are used to drive the reaction by the removal of water, and, optionally, an acylation catalyst. Esterification can also be effected using the appropriate carboxylic acid in the presence of trifluoroacetic anhydride and, optionally, pyridine, or in the presence of N,N-carbonyldiimidazole with pyridine. Reaction of an acid chloride with the alcohol can be carried out with an acylation catalyst such as 4-DMAP or pyridine. One skilled in the art would readily know how to successfully carry out these as well as other known methods of etherification of alcohols. As used herein the terms, “pharmaceutically acceptable hydrate” refer to the compounds of the instant invention crystallized with one or more molecules of water to form a hydrated form. Prodrugs and solvates of the compounds of the invention are also contemplated herein. A discussion of prodrugs is provided in T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems (1987) 14 of the A.C.S. Symposium Series, and in Bioreversible Carriers in Drug Design, (1987) Edward B. Roche, ed., American Pharmaceutical Association and Pergamon Press. The term “prodrug” means a compound (e.g., a drug precursor) that is transformed in vivo to yield a compound of the present invention or a pharmaceutically acceptable salt, hydrate or solvate of the compound. The transformation may occur by various mechanisms (e.g., by metabolic or chemical processes), such as, for example, through hydrolysis in blood. A discussion of the use of prodrugs is provided by T. Higuchi and W. Stella, “Pro-drugs as Novel Delivery Systems,” Vol. 14 of the A.C.S. Symposium Series, and in Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987. For example, if a compound of the present invention or a pharmaceutically acceptable salt, hydrate or solvate of the compound contains a carboxylic acid functional group, a prodrug can comprise an ester formed by the replacement of the hydrogen atom of the acid group with a group such as, for example, (C1-C8)alkyl, (C2-C12)alkanoyloxymethyl, 1-(alkanoyloxy)ethyl having from 4 to 9 carbon atoms, 1-methyl-1-(alkanoyloxy)-ethyl having from 5 to 10 carbon atoms, alkoxycarbonyloxymethyl having from 3 to 6 carbon atoms, 1-(alkoxycarbonyloxy)ethyl having from 4 to 7 carbon atoms, 1-methyl-1-(alkoxycarbonyloxy)ethyl having from 5 to 8 carbon atoms, N-(alkoxycarbonyl)aminomethyl having from 3 to 9 carbon atoms, 1-(N-(alkoxycarbonyl)amino)ethyl having from 4 to 10 carbon atoms, 3-phthalidyl, 4-crotonolactonyl, gamma-butyrolacton-4-yl, di-N,N—(C1-C2)alkylamino(C2-C3)alkyl (such as 3-dimethylaminoethyl), carbamoyl-(C1-C2)alkyl, N,N-di(C1-C2)alkylcarbamoyl-(C1-C2)alkyl and piperidino-, pyrrolidino- or morpholino(C2-C3)alkyl, and the like. Similarly, if a compound of the present invention contains an alcohol functional group, a prodrug can be formed by the replacement of the hydrogen atom of the alcohol group with a group such as, for example, (C1-C6)alkanoyloxymethyl, 1-((C1-C6)alkanoyloxy)ethyl, 1-methyl-1-((C1-C6)alkanoyloxy)ethyl, (C1-C6)alkoxycarbonyloxymethyl, N—(C1-C6)alkoxycarbonylaminomethyl, succinoyl, (C1-C6)alkanoyl, α-amino(C1-C4)alkanyl, arylacyl and α-aminoacyl, or α-aminoacyl-α-aminoacyl, where each α-aminoacyl group is independently selected from the naturally occurring L-amino acids, P(O)(OH)2, —P(O)(O(C1-C6)alkyl)2 or glycosyl (the radical resulting from the removal of a hydroxyl group of the hemiacetal form of a carbohydrate), and the like. If a compound of the present invention incorporates an amine functional group, a prodrug can be formed by the replacement of a hydrogen atom in the amine group with a group such as, for example, R-carbonyl, RO-carbonyl, NRR′-carbonyl where R and R′ are each independently (C1-C10)alkyl, (C3-C7)cycloalkyl, benzyl, or □-carbonyl is a natural β-aminoacyl or natural β-aminoacyl, —C(OH)COOY1 wherein Y1 is H, (C1-C6)alkyl or benzyl, —C(OY2)Y3 wherein Y2 is (C1-C4) alkyl and Y3 is (C1-C6)alkyl, carboxy (C1-C6)alkyl, amino(C1-C4)alkyl or mono-N- or di-N,N—(C1-C6)alkylaminoalkyl, —C(Y4)Y5 wherein Y4 is H or methyl and Y5 is mono-N- or di-N,N—(C1-C6)alkylamino morpholino, piperidin-1-yl or pyrrolidin-1-yl, and the like. One or more compounds of the invention may exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like, and it is intended that the invention embrace both solvated and unsolvated forms. “Solvate” means a physical association of a compound of this invention with one or more solvent molecules. This physical association involves varying degrees of ionic and covalent bonding, including hydrogen bonding. In certain instances the solvate will be capable of isolation, for example when one or more solvent molecules are incorporated in the crystal lattice of the crystalline solid. “Solvate” encompasses both solution-phase and isolatable solvates. Non-limiting examples of suitable solvates include ethanolates, methanolates, and the like. “Hydrate” is a solvate wherein the solvent molecule is H2O. One or more compounds of the invention may optionally be converted to a solvate. Preparation of solvates is generally known. Thus, for example, M. Caira et al, J. Pharmaceutical Sci., 93(3), 601-611 (2004) describe the preparation of the solvates of the antifungal fluconazole in ethyl acetate as well as from water. Similar preparations of solvates, hemisolvate, hydrates and the like are described by E. C. van Tonder et al, AAPS Pharm Sci Tech., 5(1), article 12 (2004); and A. L. Bingham et al, Chem. Commun., 603-604 (2001). A typical, non-limiting, process involves dissolving the inventive compound in desired amounts of the desired solvent (organic or water or mixtures thereof) at a higher than ambient temperature, and cooling the solution at a rate sufficient to form crystals which are then isolated by standard methods. Analytical techniques such as, for example I. R. spectroscopy, show the presence of the solvent (or water) in the crystals as a solvate (or hydrate). The compounds of the present invention have asymmetric carbon atoms and can exist in the form of stereoisomers (e.g., diastereomers, optically pure enantiomers) or as racemates or mixtures of two or more stereoisomers of each compound. The term “compound” as used herein embraces all of these forms. Diastereomers (sometimes called diastereoisomers) are stereoisomers that are not enantiomers. Diastereomerism occurs when two or more stereoisomers of a compound have different configurations at one or more (but not all) of the equivalent (related) stereocenters and are not mirror images of each other. When two diastereoisomers differ from each other at only one stereocenter they are epimers. Each stereocenter gives rise to two different configurations and thus to two different stereoisomers. Diastereomers differ from enantiomers in that the latter are pairs of stereoisomers which differ in all stereocenters and are therefore mirror images of one another. Enantiomers of a compound with more than one stereocenter are also diastereomers of the other stereoisomers of that compound that are not their mirror image. Diastereomers have different physical properties and different reactivity, unlike enantiomers. Diastereomers of the present invention include, for example (but not limited to) the two diastereomers of compounds 12, 14, 16 and 18, which differ by their stereochemistry in position C3 (steroid numbering) for example. For purposes of this Specification, “pharmaceutically acceptable tautomer” means any tautomeric form of any compound of the present invention. It is well understood by those skilled in the art that heterocyclic compounds such as the compounds of the present invention can exist under different tautomeric forms, each tautomeric form being described using a specific formula and a specific name. Herein, the use of the name or formula of one tautomeric form of a compound is meant to refer to and encompass all other tautomeric forms of the same compound. The purification of enantiomers and the separation of isomeric mixtures of a compound of the present invention may be accomplished by standard techniques known in the art. The dosages in which the compounds of the present invention are administered in effective amounts depend on the nature of the specific active ingredient, the body weight, the age and the requirements of the patient and the mode of application. In general, daily dosages of about 1 mg-5000 mg, preferably 5 mg-500 mg, per day come into consideration. The skilled artisan will appreciate that certain factors may influence the dosage required to effectively treat a subject, including but not limited to the severity of the disease or disorder, previous treatments, the general health and/or age of the subject, and other diseases present. Moreover, treatment of a subject with a therapeutically effective amount of a compound of the present invention can include a series of treatments. Toxicity and therapeutic efficacy of such compounds can be determined by standard pharmaceutical procedures in cell cultures or experimental animals, e.g., for determining the LD50(the dose lethal to 50% of the population) and the ED50(the dose therapeutically effective in 50% of the population). The dose ratio between toxic and therapeutic effects is the therapeutic index and it can be expressed as the ratio LD50/ED50. Compounds that exhibit large therapeutic indices are preferred. While compounds that exhibit toxic side effects may be used, care should be taken to design a delivery system that targets such compounds to the site of affected tissue in order to minimize potential damage to uninfected cells and, thereby, reduce side effects. Data obtained from cell culture assays and animal studies can be used in formulating a range of dosage for use in humans. The dosage of such compounds lies preferably within a range of circulating concentrations that include the ED50with little or no toxicity. The dosage may vary within this range depending upon the dosage form employed and the route of administration utilized. For any compound used in the method of the invention, the therapeutically effective dose can be estimated initially from cell culture assays. A dose may be formulated in animal models to achieve a circulating plasma concentration range that includes the IC50(i.e., the concentration of the test compound which achieves a half-maximal inhibition of symptoms) as determined in cell culture. Such information can be used to more accurately determine useful doses in humans. Levels in plasma may be measured, for example, by high performance liquid chromatography. The present invention also encompasses kits comprising the compounds of the present invention. For example, the kit can comprise one or more compounds inhibiting the growth of electron transport-deficient microbes (e.g., SCVs) or potentiating the antimicrobial activity of aminoglycoside antibiotics against normal bacterial strains (e.g., staphylococci). The kit may optionally include one or more control sample(s). The compounds or agents can be packaged in a suitable container. The kit can further comprise instructions for using the kit. The present invention also relates to methods for preparing the above-mentioned compounds. The present invention is illustrated in further details by the following non-limiting examples. All moisture-sensitive reactions were performed in an inert, dry atmosphere of argon in flame-dried glassware. Air-sensitive liquids were transferred via syringe or cannula through rubber septa. Reagent grade solvents were used for extractions and flash chromatography. THF was distilled from sodium/benzophenone under argon; methanol was distilled from magnesium under argon. All other solvents purchased from commercial sources were used without further purification. Tomatidine hydrochloride (3) was purchased from Molekula. All other reagents were generally purchased from Aldrich and used with no further purification. Reactions were monitored by analytical thin-layer chromatography (Canadian Life Science, TLC, GLASS plates SIL 60 G-25 UV 254). The plates were visualized first with UV illumination followed by heating with ceric ammonium molybdate (CAM) [1% (w/v) ammonium cerium sulphate, 2.5% (w/v) molybdenum trioxide, 1:9 H2SO4/H2O]. Flash column chromatography was performed manually on SiliaFlash® P60 silica. The solvent compositions reported for all chromatographic separations are on a volume/volume (v/v) basis. High-pressure liquid chromatography (HPLC) was performed on an Agilent 1100™ apparatus using an ACE C18 column, 250×21.2 mm, with 5 μm silica and 15.5% carbon load. NMR spectra were recorded at room temperature on a Bruker Spectrospin 300™ spectrometer at 300 MHz (1H) and 75 MHz (13C), or on a Varian Unity Inova™ at 600 HMz (1H) and 150 MHz (13C).1H NMR spectra are reported in parts per million (ppm) on the δ scale relative to the residual signals of chloroform (δ 7.26 ppm) or methanol (δ 3.31 ppm) as an internal standard with the following multiplicity—s: singlet; d: doublet; t: triplet; q: quartet; quint: quintet; m: multiplet.).13C NMR spectra are reported in ppm on the δ scale relative to CDCl3(δ 77.0 ppm) or CD3OD (δ 49.0 ppm). High-resolution mass spectrometry (HRMS) was performed on a Maxis (Bruker) Q-TOF. Boc-alkanediamines 38 and 40 were synthesized as reported by Jensen et al. Reductive amination with pregnanolone acetate derivatives were synthesized as reported by Xie et al, 2001. In a 25 mL round bottom flash equipped with a condenser, the starting material was solubilized in 5 mL anhydrous MeOH. To this solution was added a solution of 0.9 mL acetyl chloride in 6 mL anhydrous MeOH. The reaction heated to reflux for 1 h, monitored by TLC. The reaction was allowed to cool at room temperature, then the solvent was removed in vacuo to yield the desired compound. In a 250 mL round flask, tomatidine hydrochloride (3) (200 mg, 0.44 mmol, 1.0 eq) was suspended in dry THF (20 mL) then acetic formic anhydride (380 mg, 4.42 mmol, 10.0 eq) was added, followed by DIPEA (390 μL, 2.20 mmol, 5.0 eq). The reaction was stirred for 15 minutes, then monitored by TLC (50% EtOAc/Hexanes, CAM stain, Rf=0.23 for mono-formylated compound, 0.49 for diformylated compound). The volatiles were removed by evaporation under reduced pressure. The compound was then diluted in a mixture of 125 mL EtOH and 50 mL aqueous NaHCO3buffer (pH=9.5) and stirred for one week, monitored by TLC until the complete disappearance of the diformylated compound. EtOH was then evaporated, and the resulting aqueous phase was extracted with EtOAc (3×25 mL). The combined organic phases were dried on anhydrous magnesium sulphate then evaporated under reduced pressure. The crude product was purified by flash chromatography (25% EtOAc/Hexanes) to give 155 mg (79%) of the desired compound 4 as a white solid (m.p. 184-187° C.). 1H NMR (600 MHz, CDCl3) δ (ppm) 8.41 (s, 1H), 4.29 (d, 1H, J=11.5 Hz), 4.13 (dd., 1H, J1=7.3 Hz, J2=15.5 Hz), 3.58 (quint, 1H, J=4.7 Hz), 2.65 (t, 1H, J=11.5), 2.54 (quint, 1H, J=7.1 Hz), 1.98 (quint, 1H, J=5.28 Hz) 1.87 (d, 1H, J=13.7 Hz), 1.82-1.72 (m, 3H), 1.72-1.63 (m, 3H), 1.61-1.45 (m, 7H), 1.40 (d, 1H, J=13.0 Hz), 1.38-1.22 (m, 8H), 1.15 (dt, 1H, J1=12.3 Hz, J2=3.9 Hz), 1.12-1.06 (m, 2H), 1.05 (d, 3H, J=6.8 Hz), 0.95 (dt, 1H, J1=13.7 Hz, J2=3.6 Hz), 0.91 (d, 3H, J=5.9 Hz), 0.89-0.84 (m, 1H), 0.82 (s, 6H), 0.64 (dt, 1H, J1=11.39, J2=3.6 Hz). 13C NMR (75.5 MHz, CDCl3) δ (ppm) 158.9, 97.8, 78.5, 72.1, 62.3, 55.8, 54.3, 44.8, 40.2, 38.1, 36.9, 36.8, 35.5, 35.0, 32.2, 31.4, 30.7, 29.7, 29.2, 28.5, 27.5, 21.0, 18.9, 17.0, 15.3, 12.3. HRMS calculated for C28H45O3NNa: 466.3292. found: 466.3308 (MNa+). In a 10 mL round bottom flask, N-formyltomatidine 4 (50 mg, 0.113 mmol, 1.0 eq) and Dess-Martin periodinane (95 mg, 0.225 mmol, 2.0 eq) were stirred in 6.5 mL DCM. The reaction was monitored by TLC (50% AcOEt/Hexanes, Rf: 0.24). Upon completion, the reaction was quenched for 30 minutes with Na2S2O3(0.2M), then extracted 3× with EtOAc. The combined organic phases were washed with brine, dried on anhydrous magnesium sulphate then evaporated under reduced pressure. The crude compound was purified by flash chromatography (50% EtOAc/Hexanes) to yield 34 mg (68%) of the desired compound. 1H NMR (300 MHz, CDCl3) δ (ppm) 8.46 ppm (s, 1H), 4.32 (d, 1H, J=12.8 Hz), 4.17 (quad, 1H, J=8.9 Hz), 2.69 (t, 1H, J=12.6 Hz), 2.58 (quint, 1H, J=6.6 Hz), 2.52-2.24 (m, 3H), 2.16-2.11 (m, 1H), 2.11-1.98 (m, 3H), 1.91 (d, 1H, J=14.0 Hz), 1.86-1.68 (m, 4H), 1.69-1.42 (m, 7H), 1.40-1.20 (m, 7H), 1.19-1.12 (m, 2H), 1.09 (d, 3H. J=7.1 Hz), 1.05 (s, 2H), 0.94 (d, 4H, J=5.5 Hz), 0.88 (s, 3H), 0.77 (dt, 1H, J1=12.6 Hz, J2=4.8 Hz). In a 25 mL round flask, 44 mg N-formyl-3-oxotomatidine (9) (0.1 mmol) was dissolved in methanol (6 mL) along with ammonium acetate (NH4OAc) (77 mg, 1.0 mmol, 10 eq). The pH was adjusted to 6 with acetic acid, sodium cyanoborohydride (NaBH3CN) (6.9 mg, 0.11 mmol, 1.1 eq) was added and the reaction was refluxed overnight until complete as monitored by TLC (10% MeOH/AcOEt with 0.5% NEt3, Rf: 0). Solvents were removed under reduced pressure, and the solid was suspended in water. The pH was adjusted to 8 with saturated aqueous NaHCO3. The mixture was extracted with 3× EtOAc, and the combined organic fractions were washed with brine, dried on anhydrous magnesium sulphate and evaporated under reduced pressure. The crude compound was purified by flash chromatography (10% MeOH/89% EtOAc/1% NEt3) to yield 21 mg (48%) of the desired compound 11. 1H NMR (300 MHz, CDCl3) δ (ppm) 8.40 (s, 1H), 5.91, (broad s, 2H), 4.28 (d, 1H, J=11.5 Hz), 4.12 (m., 1H), 2.86 (quint, 1H, J=7.1 Hz), 2.65 (t, 1H, J=11.5), 2.53 (quint, 1H, J=7.1 Hz), 2.04-1.92 (m, 3H) 1.90-1.82 (m, 1H), 1.80-1.42 (m, 11H), 1.38-1.18 (m, 9H) 1.17-1.06 (m, 2H), 1.04 (d, 3H, J=7.1 Hz), 0.90 (d, 4H, J=6.2 Hz), 0.81 (s, 6H), 0.69-0.57 (m, 1H). Following procedure A, 770 μL diaminoethane was used to yield 76 mg (41%) of compound 38 as a colorless oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 5.28-5.11 (broad s, 1H), 3.13 (q, 2H, J=5.7 Hz), 2.76 (t, 2H, J=5.7 Hz), 2.47-2.20 (broad s, 2H), 1.38 (s, 9H). Following the procedure used for the synthesis of compound 11, 110 mg 9 (0.25 mmol), compound 38 (BocNH(CH2)2NH2) (200 mg, 1.25 mmol, 5 eq) and sodium cyanoborohydride (NaBH3CN) (17 mg, 0.27 mmol, 1.1 eq) were used to yield 130 mg (85%) of compound 13. (RF: 0.05 in 10% MeOH/AcOEt with 0.5% NEt3)1HNMR shows the presence of residual starting diamine, which was removed in the next step. 1H NMR (300 MHz, CDCl3) δ (ppm) 8.39 (s, 1H), 5.02, (s large, 1H), 4.28 (d, 1H, J=11.5 Hz), 4.12 (m, 1H), 3.24-3.15 (m, 2H), 2.77-2.69 (m, 2H), 2.64 (m, 1H) 2.54 (m, 1H), 2.44 (m, 1H), 2.04-1.46 (m, 18H) 1.43 (s, 12H), 1.33-1.15 (m, 14H), 1.04 (d, 3H, J=6.9 Hz), 0.90 (d, 3H, J=5.7 Hz), 0.87-0.82 (m, 2H), 0.81 (s, 3H), 0.78 (s, 3H), 0.69-0.57 (m, 1H). Following procedure D, 130 mg of compound 13 were deprotected in quantitative yield (Rf: 0 in 10% MeOH/AcOEt with 0.5% NEt3). The crude compound was purified preparative HPLC to yield 48 mg (38%) of desired compound 14. The HPLC elution parameters were as follow (percentage of acetonitrile in water): 0 min, 20%; 2 min, 20%; 27 min, 95%; 32 min, 95%. 1H NMR (300 MHz, CD3OD) δ (ppm) 4.36 (m, 1H), 3.18-3.03 (m, 2H), 2.89 (t, 1H, J=10.7 Hz) 2.20 (m, 1H), 2.08-1.90 (m, 3H), 1.80-1.50 (m, 18H), 1.45-1.14 (m, 12H) 1.09 (d, 3H, J=7.1 Hz), 0.69 (d, 3H, J=6.7 Hz), 0.89 (s, 3H), 0.85 (s, 4H), 0.83-0.66 (m, 2H). 13C NMR (75.5 MHz, CD3OD) δ (ppm) 96.1, 81.1, 61.6, 57.6, 56.3, 55.4, 53.7, 44.5, 41.1, 40.8, 40.7, 39.5, 38.6, 35.7, 35.3, 34.8, 31.7, 31.4, 30.7, 28.9, 28.0, 25.8, 25.3, 24.4, 20.6, 17.2, 15.9, 11.1, 10.4. HRMS calculated for C29H52ON3+: 458.4105. found: 458.4105 (MH+). Following procedure A, 1.16 mL diaminobutane was used to yield 171 mg (79%) of compound 40 as a colorless oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 4.94 (broad s, 1H), 3.04 (broad s, 4H), 2.69 (broad s, 2H), 1.45 (broad s, 4H), 1.36 (s, 9H). Following the procedure used for the synthesis of compound 11, 27 mg 9 (0.060 mmol), compound 40 (BocNH(CH2)4NH2) (57 mg, 0.30 mmol, 5 eq) and sodium cyanoborohydride (NaBH3CN) (4.1 mg, 0.066 mmol, 1.1 eq) were used to yield 16 mg (44%) of compound 15. (Rf: 0.07 in 10% MeOH/AcOEt with 0.5% NEt3) 1H NMR (300 MHz, CDCl3) δ (ppm) 8.40 (s, 1H), 5.02, (broad d, 1H), 4.28 (d, 1H, J=11.5 Hz), 4.12 (m, 1H), 3.14-3.04 (s large, 3H), 2.80-2.50 (m, 5H), 2.42 (m, 1H), 2.04-1.93 (m, 2H), 1.86 (d, 1H, J=12.8 Hz), 1.79-1.62 (m, 5H), 1.60-1.46 (m, 12H), 1.42 (s, 12H), 1.35-1.10 (m, 10H), 1.04 (d, 5H, J=6.9 Hz), 0.90 (d, 4H, J=5.8 Hz), 0.81 (s, 3H), 0.78 (s, 3H), 0.69-0.57 (m, 1H). Following procedure D, 16 mg of compound 15 were deprotected in quantitative yield. (RF: 0 in 10% MeOH/AcOEt with traces if NEt3) The crude compound was purified by preparative HPLC to yield 15 mg (100%) of desired compound 16. The HPLC elution parameters were as follow (percentage of acetonitrile in water): 0 min, 5%; 2 min, 5%; 27 min, 95%; 32 min, 95%. 1H NMR (300 MHz, CD3OD) δ (ppm) 4.37 (q, 1H, J=4.8 Hz), 3.38 (broad s, 1H), 3.18-3.15-2.87 (m, 7H), 2.20 (t, 1H, J=6.5 Hz), 2.08-1.82 (m, 6H) 1.82-1.63 (m, 11H), 1.61-1.50 (m, 5H), 1.44-1.14 (m, 12H), 1.08 (d, 4H, J=7.4 Hz), 0.96 (d, 3H, J=6.5 Hz), 0.89 (s, 3H), 0.87 (s, 4H), 0.79-0.69 (m, 1H). 13C NMR (75.5 MHz, CD3OD) δ (ppm) 96.5, 80.7, 61.8, 57.1, 55.4, 54.9, 53.8, 45.2, 44.6, 43.6, 41.0, 39.6, 38.6, 36.2, 35.3, 34.8, 31.7, 31.5, 30.8, 29.1, 28.5, 28.0, 26.1, 25.4, 24.5, 24.3, 22.7, 22.3, 20.6, 17.4, 15.9, 13.4, 11.0. HRMS calculated for C31H57ON32+: 243.7245. found: 243.7253 Following the procedure used for the synthesis of compound 11, 20 mg 9 (0.045 mmol), hexamethylenediamine (BocNH(CH2)6NH2) (30 μL, 0.225 mmol, 5 eq) and sodium cyanoborohydride (NaBH3CN) (3.2 mg, 0.050 mmol, 1.1 eq) were used to yield 11 mg (46%) of compound 17 (Rf: 0 in 10% MeOH/AcOEt with 0.5% NEt3). 1H NMR (300 MHz, CDCl3) δ (ppm) 4.32-4.25 (m, 1H), 4.18-4.08, (m, 1H), 3.08 (broad s, 3H), 2.74-2.49 (m, 5H), 2.03-1.93 (m, 2H) 1.91-1.81 (m, 1H), 1.80-1.70 (m, 2H), 1.70-1.41 (m, 11H), 1.39-1.09 (m, 14H), 1.05 (d, 3H, J=7.0 Hz), 0.90 (d, 3H, J=5.4 Hz), 0.87-0.82 (m, 2H), 0.82 (s, 3H), 0.80 (s, 3H), 0.69-0.59 (m, 1H). Following procedure D, 11 mg of compound 17 were deprotected in quantitative yield (Rf: 0 in 10% MeOH/AcOEt with 0.5% NEt3). The crude compound was purified by preparative HPLC to yield 12.5 mg (100%) of compound 18. The HPLC elution parameters were as follow (percentage of acetonitrile in water): 0 min, 20%; 2 min, 20%; 27 min, 95%; 32 min, 95%. 1H NMR (300 MHz, CD3OD) δ (ppm) 4.37 (m, 1H), 3.39 (m, 1H), 3.16-3.05 (m, 1H) 3.04-2.82 (m, 6H), 2.20 (t, 1H, J=6.7 Hz), 2.08-1.95 (m, 3H), 1.90-1.82 (m, 3H), 1.80-1.52 (m, 15H), 1.50-1.16 (m, 18H), 1.09 (d, 3H, J=6.4 Hz), 1.06-0.99 (m, 3H), 0.96 (d, 3H, J=6.8 Hz), 0.90 (s, 3H), 0.88 (s, 4H), 0.75-0.60 (m, 1H). 13C NMR (75.5 MHz, CD3OD) δ (ppm) 96.1, 81.0, 61.6, 57.0, 55.5, 53.8, 53.5, 44.5, 44.2, 40.8, 39.5, 38.8, 35.6, 34.7, 31.5, 31.3, 31.2, 30.8, 29.0, 28.2, 27.6, 26.9, 25.9, 25.5, 25.4, 22.2, 20.2, 17.2, 15.8, 13.1, 10.3. HRMS calculated for C33H61ON3+: 257.7402. found: 257.7406. Following procedure B, 50 mg of compound 28 (0.14 mmol), 137 μL (1.39 mmol, 10 eq) of butylamine and 10 mg of sodium cyanoborohydride (0.15 mmol, 1.1 eq) were converted to 24.2 mg (41%) of desired product (Rf: 0.33 in 10% MeOH/AcOEt with 0.5% NEt3). 1H NMR (300 MHz, CDCl3) δ (ppm) 4.67 (sept, 1H, J=5.9 Hz), 2.75-2.65 (m, 1H), 2.60-2.52 (m, 1H), 2.50-2.41 (m, 1H), 2.01 (s, 3H), 2.00-1.84 (m, 5H), 1.82-1.44 (m, 11H, 1.40-1.10 (m, 12H), 1.09 (d, 5H, J=6.5 Hz), 1.07-0.95 (m, 3H), 0.91 (t, 3H, J=7.4 Hz), 0.81 (s, 3H), 0.65 (s, 4H). Following procedure D, 15 mg of compound 47 (0.036 mmol), were converted to 13.5 mg (100%) of compound 48 (Rf: 0.05 in 10% MeOH/AcOEt with 0.5% NEt3). 1H NMR (300 MHz, CD3OD) δ (ppm) 3.48 (m, 1H), 3.20 (m, 1H), 2.97 (m, 2H), 1.97-1.84 (m, 2H), 1.79-1.07 (m, 23H), 1.01-0.85 (m, 5H), 0.81 (s, 3H), 0.72 (s, 3H), 0.69-0.61 (m, 1H). 13C NMR (75.5 MHz, CD3OD) δ (ppm) 70.4, 57.4, 56.0, 54.1, 52.7, 44.7, 42.7, 42.6, 39.1, 37.4, 36.8, 35.2, 35.2, 31.7, 30.7, 28.4, 27.9, 25.8, 23.8, 20.8, 19.5, 15.1, 12.5, 11.3, 10.9. HRMS calculated for C25H46ON: 376.3574. found: 376.3578 (MH+). Following procedure B, 100 mg of compound 28 (0.28 mmol), 200 μL (1.72 mmol, 6.2 eq) of isopentylamine and 18 mg of sodium cyanoborohydride (0.29 mmol, 1.05 eq) were converted to 46 mg (41%) of compound 49 (Rf: 0.36 in 10% MeOH/AcOEt with 0.5% NEt3). 1H NMR (300 MHz, CDCl3) δ (ppm) 5.62 (s large, 2H), 4.65 (sept, 1H, J=6.2 Hz), 3.26-2.57 (m, 2H), 1.99 (s, 5H), 1.85-1.37 (m, 11H), 1.35-1.15 (m, 8H), 1.12 (d, 2H, J=6.3 Hz), 1.09-0.96 (m, 3H), 0.92 (d, 1H, J=6.3 Hz), 0.89 (d, 6H, J=6.4 Hz), 0.790 (s 3H), 0.69-0.55 (m, 4H). Following procedure D, 46 mg of compound 49 (0.11 mmol), were converted to 48 mg (100%) of compound 50 (Rf: 0.07 in 10% MeOH/AcOEt with 0.5% NEt3). 1H NMR (300 MHz, CD3OD) δ (ppm) 3.49 (m, 1H), 2.98 (m, 1H), 1.89-1.45 (m, 12H), 1.45-1.00 (m, 17H), 0.96 (d, 6H, J=6.4 Hz), 0.91-0.86 (m, 2H), 0.82 (s, 3H), 0.72 (s, 3H), 0.70-0.63 (m, 2H). 13C NMR (75.5 MHz, CD3OD) δ (ppm) 70.4, 55.6, 54.4, 54.0, 52.0, 44.8, 42.3, 37.5, 37.4, 36.8, 35.2, 34.4, 31.7, 30.7, 28.4, 25.9, 23.5, 23.4, 21.4, 21.2, 20.6, 14.3, 11.5, 11.3. HRMS calculated for C26H48ON+: 390.3730. found: 390.3733 (MH+). The syntheses of compounds described in Examples 1 to 15 are also presented in Method. The minimal inhibitory concentrations (MICs), (i.e. lowest concentration of an antimicrobial that will inhibit the visible growth of a microorganism after incubation), of test compounds were determined against the normal or SCV strains of bacterial species from the Firmicutes phylum. Briefly, MICs were determined using the microdilution method in 96-well plates (Clinical and Laboratory Standards Institute (CLSI), 2011). Bacteria were inoculated at 105-106CFU/mL and incubated at 35° C. for 20 h (normal strains) or 48 h (SCVs) in brain heart infusion (BHI) broth in order to allow SCVs to reach maximal growth as previously described (Mitchell et al., 2012). Then OD595 nmwas read on a microplate reader. The MICs obtained against the quality control strain Results. The various antibacterial activities of the test compounds against Interestingly, certain compounds, notably compounds 14, 16 and 18 showed some antibacterial activities of their own against Also in addition to that seen in Table 1 with gentamicin, Table 2 reports the activity of Compound 14 in combination with a variety of antibiotics of the aminoglycoside class showing that a combination with any aminoglycoside leads to an important synergy against a normal The potentiation effects of Compound 14 against antibiotic resistant strains such as methicillin-resistant Results: Table 3 reports the antibacterial activity of Compound 14 on MRSA strains (MIC of 16 μg/ml when used alone). This activity is similar to that determined against the methicillin-susceptible strain Time-kill experiments were performed in order to determine whether the effect of test compounds or the combination with an aminoglycoside on Method: Bacteria were inoculated at 105-106CFU/ml in BHI in the absence or presence of antibiotics at the specified concentrations: concentrations of 8 μg/ml of tomatidine hydrochloride salt (TO); 8 μg/ml of Compound 14 (Cpd); 2 μg/ml of gentamicin (GEN); combination of 8 μg/ml TO and 2 μg/ml gentamicin; or combination of 8 μg/ml Compound 14 and 2 μg/ml gentamicin ( Results: Time-kill experiments were performed in order to determine whether the effect of test compounds on SCVs of bacterial species of the Firmicutes phylum is bacteriostatic (prevents growth) or bactericidal (kills cells). Method: One of the Results: The bactericidal activity of tomatidine and Compound 14 in combination with gentamicin against Method: Results: The biological activity of compounds of the present invention, used alone, can be evaluated using techniques as described in Examples 20-23 to determine the antibacterial activity against SCV strains (e.g., Also determined is the antibacterial activity against additional Firmicutes of the Bacillales group such as coagulase-positive and -negative staphylococci including The compounds of the present invention were tested for their ability to inhibit the growth of normal bacteria alone or in combination with aminoglycosides or of bacteria with electron transport deficiencies during infection of cell cultures. Results herein show that Compound 14 has an antimicrobial activity against intracellular SCVs. This is particularly relevant because the ability of SCVs to persist within host cells is thought to be involved in the development of chronic and difficult-to-treat infections (Sendi and Proctor, 2009). This is also of particular relevance because Method: Human airway epithelial cells, the Calu-3 cell line ATCC HTB 55, were used in this cell culture assay. Cells were cultured in Eagle's Minimum Essential Medium (EMEM) supplemented with 0.1 mM MEM nonessential amino acids, 1 mM of sodium pyruvate, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 2.5 μg/ml of Fungizone and 10% fetal bovine serum (FBS) at 37° C. in 5% CO2. For routine culture, 4 μg/ml of puromycin was added to culture media. All cell culture reagents were purchased from Wisent (St-Bruno, QC, Canada). Cell infection assays were performed as previously described (Mitchell et al., 2011). Cells were seeded at 2.5×105cells/inserts on 12-well Transwell™ plates and cultured for 9 to 10 days in an air:liquid system. The complete medium in the basal compartment was replaced by the invasion medium (1% FBS and no antibiotics) 18 h before assays. Inocula were prepared by suspending bacteria grown 20 h on BHIA plates in ice-cold PBS. Bacteria (the Results: Tomatidine was used as a selective pressure to raise tomatidine-resistant Method: The Illumina sequencing libraries were prepared using the multiplexing method of Rodrigue et al., 2010. Briefly, genomic DNA from bacteria were sheared by sonication using a Bioruptor Plus™, and the resulting DNA molecules were treated with T4 DNA polymerase and T4 polynucleotide kinase to be made blunt and phosphorylated. AMPure™ XP SPRI beads were used for size selection of the DNA fragments at an average size of ˜500 bp. Short double-stranded adaptors were ligated and subjected to a nick translation reaction using Taq DNA polymerase. The adaptors were used as priming sites for PCR amplification to incorporate a 6 bp index for sample multiplexing. The size and concentration of the libraries were determined before being mixed and sent for sequencing on the Illumina HiSeq2000™ platform at the Genome Quebec and McGill University Innovation Centre (Montreal, QC, Canada). The obtained sequences were then aligned against reference Results: Tomatidine-resistant mutants were found in the bacterial colonies isolated after sequential passages on tomatidine and on tomatidine and gentamicin. Genomic mutations that were conserved in each of the three isolated colonies of each of the passages showing the same level of resistance to tomatidine (MIC>32 ug/mL) were associated to the molecular target. The target of tomatidine was thus identified as the ATP synthase subunit c (gene NWMN_2012, from the The bacterial ATP synthase (also known as F-ATPase, F-Type ATPase or FOF1-ATPase) is divided into two regions: a cytoplasmic globular F1catalytic sector and a membrane-bound FOproton-translocating sector (Hong and Pedersen, 2008). F1(composed of five subunits: α3β3γδε) generates ATP from proton translocation through FO. FOis composed of three types of subunits, namely subunit a, subunit b and subunit c. FOcontains 1 subunit a, 2 subunits b and 8 to 15 subunits C, which is denoted by ab2c8-15. Compounds of the present invention target the c subunits (c-ring) of FO. The “O” in FOcomes from oligomycin. Oligomycin binds at the interface of subunit a and the c-ring oligomer and is a poison that specifically blocks proton transport through FOin the mitochondrial ATP synthase. Oligomycin is mostly ineffective on ATP synthases from most bacteria. Inversely, Bedaquiline (TMC207) is specific for the Mycobacterial ATP synthase (i.e., has no inhibitory activity against Two different mutations were found in the ATP synthase gene after sequential passages of the Tomatidine and Compound 14 MICs were determined for As in Example 27 with tomatidine, Compound 14 is used as a selective pressure to raise Compound 14-resistant Method: A large inoculum (108-1010bacteria) of the The effect of test compounds on the generation of ATP by the ATP synthase of inverted membrane vesicles of The compounds of the present invention are able to inhibit the growth of normal bacteria alone or in combination with aminoglycosides or of bacteria with electron transport deficiencies during infection of an animal (in vivo). The antibacterial activity in vivo is demonstrated through the use of various infection models using, for example mice models of septicemia, soft tissue infections, respiratory infections, burned tissues, and mastitis. The compounds of the present invention are able to inhibit the growth of mixed bacterial species, such as The septicemia model (Deslouches et al, 2005) allows testing the efficacy of compounds to clear or diminish an infection. Bacteria are injected iv or ip with an inoculum that leads to 50-70% mortality in untreated mice (3-5 mice per test group). Following inoculation, compounds are administered either iv, ip, sc or im and treatment efficacy is measured by the reduction of bacterial CFU in various organs (e.g., liver, kidneys), in the peritoneal liquid or in blood or is evaluated based on the animals' survival rate. Compound efficacy in a neutropenic mouse thigh model is evaluated as follows (Malouin et al, 2005): Mice (immune suppressed with cyclophosphamide treatments prior to infection) are challenged with bacteria (104CFU per thigh im). To determine efficacy, compounds are delivered iv, sc, ip or im 2 h post-infection. Mice (3-5 mice per treatment) are euthanized 8 h post-infection. The thigh tissues (two samples per animal) are recovered, homogenized, and bacterial CFU per g of tissue are determined by plating appropriate dilutions. Compound efficacy in a lung infection (pneumonia) model was evaluated as follow (Ragle et al, 2010; Mitchell et al, 2013): Mice (36) were challenged with an intra-tracheal injection of Results are presented in This shows that Compound 14 can significantly reduce the bacterial load in mouse lungs infected with A burned tissue model is performed as follow. Female CF-1 mice (22 to 25 g) receive a nonlethal, partial-thickness alcohol flame burn (10 s) covering 15% of the body surface (Goldberg et al., 1995) or by causing the thermal injury to the skin by placing the shaved exposed back area to 90° C. water for 10 s (McVay et al., 2007). Immediately after the thermal injury, the animals receive 0.5 ml of sterile normal saline subcutaneously as fluid replacement therapy. Subsequently, bacteria (102CFU) such as Compound efficacy in a mouse mastitis model is evaluated as follow (Brouillette et al, 2004b): Lactating CD-1 mice are challenged with bacteria injected through the teat canal. A Hamilton syringe with a blunt needle is used to inoculate with 102CFU per gland in both L4 and R4 mammary glands. Compounds are delivered by an intra-mammary injection 4 h following challenge. Each experimental group is composed of 3-6 mice (i.e., 6-12 glands). Mammary glands are harvested, weighed and homogenized in PBS at 18 h. Homogenates are serially diluted and plated on agar for bacterial CFU determination. Although the present invention has been described hereinabove by way of specific embodiments thereof, it can be modified, without departing from the spirit and nature of the subject invention as defined in the appended claims. The present invention provides bacterial ATP synthase inhibitors such as a compound of formula (I): (Formula (I)) (A) in combination with an aminoglycoside antibiotic for preventing or treating a bacterial infection in a subject; or (B) (a) for preventing or treating an infection caused by an electron transport-deficient bacteria in a subject; or (b) for the disinfection, sterilization and/or antisepsis of an object contaminated with an electron transport-deficient bacteria. There are provided compositions and kits using such compounds and inhibitors. 1. A method of using a compound of formula (I): wherein: R1 is H; and R2 is —OH or —NR5R6; or R1 is —OH or —NR5R6; and R2 is H;
wherein R5 and R6 are identical or different and are selected from the group consisting of H; substituted or unsubstituted alkyl; substituted or unsubstituted aryl; substituted or unsubstituted aralkyl; substituted or unsubstituted cycloalkyl, and substituted or unsubstituted —(CH2)nNR7R8,
wherein n is 2-10; and R7 and R8 are identical or different and are selected from the group consisting of H, substituted or unsubstituted alkyl, substituted or unsubstituted aryl; substituted or unsubstituted cycloalkyl, and substituted or unsubstituted aralkyl; wherein the substituted alkyl(s), substituted aryl(s), substituted aralkyl(s), substituted cycloalkyl(s), and substituted —(CH2)nNR7R8 comprise(s) one or more identical or different substitutions selected from alkyl, aryl, cycloalkyl and aralkyl; R3 is —C(CH3)(NHR9),
wherein R9 is H, substituted or unsubstituted alkyl, or substituted or unsubstituted —(CH2)mNR11R12, wherein m is 2-10; and
R11 and R12 are identical or different and are selected from the group consisting of H, substituted or unsubstituted alkyl, substituted or unsubstituted aryl, substituted or unsubstituted cycloalkyl, and substituted or unsubstituted aralkyl wherein the substituted alkyl(s), substituted aryl(s), substituted aralkyl(s), substituted cycloalkyl(s), and substituted —(CH2)mNR11R12 comprise(s) one or more identical or different substitutions selected from alkyl, aryl, cycloalkyl and aralkyl; and R4 is H,
with the proviso that when R3 is —C(CH3)(NHR9), R4 is H, R1 is OH and R2 is H, R9 is substituted or unsubstituted alkyl; wherein the substituted alkyl comprises one or more identical or different substitutions selected from alkyl, aryl, cycloalkyl and aralkyl; or R3 and R4 form together with the carbon atoms to which they are attached form a spirocyclic structure of formula II: wherein R10 is H, substituted or unsubstituted alkyl, or substituted or unsubstituted —(CH2)pNR13R14,
wherein p is 2-10; and R13 and R14 are identical or different and are selected from the group consisting of H, substituted or unsubstituted alkyl, substituted or unsubstituted aryl, substituted or unsubstituted cycloalkyl, and substituted or unsubstituted aralkyl; wherein substituted alkyl, substituted aryl, substituted aralkyl, substituted cycloalkyl, and substituted —(CH2)pNR13R14 comprises one or more identical or different substitutions selected from alkyl, aryl, cycloalkyl and aralkyl, with the proviso that when R3 and R4 form together a spirocyclic structure of formula II, neither R1 nor R2 is —OH; and with the proviso that when R1 is NH2 and R2 is H; or R1 is H and R2 is NH2, R10 is not H, or a stereoisomer or mixture of stereoisomers or a salt thereof, (A) in combination with an aminoglycoside antibiotic for preventing or treating a bacterial infection in a subject; or (B) (a) for preventing or treating an infection caused by an electron transport-deficient bacteria in a subject; or (b) for the disinfection, sterilization and/or antisepsis of an object contaminated with an electron transport-deficient bacteria. 2. The method of 3. The method of 4. The method of 5. The method of 6. (canceled) 7. (canceled) 8. (canceled) 9. (canceled) 10. (canceled) 11. The method of 12. The method of 13. The method of 14. The method of or
(ii) or a salt, stereoisomer or any mixture of stereoisomers of (i) or (ii). 15. The method of 16. The method of 17. (canceled) 18. (canceled) 19. The method of 20. The method of 21. The method of 22. (canceled) 23. (canceled) 24. The method of 25. (canceled) 26. (canceled) 27. (canceled) 28. The method of (ii) or
(iii) or a salt, stereoisomer or any mixture of stereoisomers of any one of (i) to (iii). 29. The method as defined in 30. (canceled) 31. The method of 32. (canceled) 33. The method of any one of 34. (canceled) 35. The method of 36. The method of 37. (canceled) 38. The method of 39. (canceled) 40. (canceled) 41. The compound as defined in 42. A method of using a compound inhibiting a bacterial ATP synthase by targeting the ATP synthase subunit C conserved region:
(i) LX1X2X3AAAIAX4GLX5ALGAGIGNGLIVX6X7TX8EGX9ARQPEX10as set forth in SEQ ID NO: 24, 25 or 26; or (ii) GZ1Z2LVEALPIIZ3VVIAF as set forth in SEQ ID NO: 30, 31 or 32, (A) in combination with an aminoglycoside antibiotic for preventing or treating an infection by a Firmicutes phylum bacteria of the order Bacillales in a subject; or (B) (a) for preventing or treating a bacterial infection caused by an electron transport-deficient bacteria in a subject; or (b) for the disinfection, sterilization and/or antisepsis of an object contaminated with an electron transport-deficient bacteria, wherein the inhibitor is not tomatidine, tomatidine 3-sulfate, tomatidine 3-phosphate, 3α-hydroxytomatidine, 3-oxo-tomatidine, 3-aminotomatidine, N-formyl tomatidine, N-formyl-3α-acetyltomatidine, 3α-hydroxytomatidine, N-formyl-3-oxotomatidine, 3-oxotomatidine, O-allyl-N-formyltomatidine, O-allyltomatidine, tomatidine methanesulfonate, tomatidine citrate, O-acetyl-N-benzylpregn-5,6-en-3β-ol-20-amine, pregnan-3β-ol-20-amine, O-acetylpregn-5,6-en-3β-ol-20-((N, N-dimethylamino)propyl)amine, pregnan-3β-ol-20-((N,N-dimethylamino)propyl)amine, O-acetylpregnan-3β-ol-20-(boc-aminoethyl)amine, O-acetylpregnan-3β-ol-20-(boc-aminopropyl)amine, O-acetylpregnan-3β-ol-20-(boc-aminobutyl)amine, pregnan-3β-ol-20-(boc-aminoethyl)amine, pregnan-3β-ol-20-(boc-aminopropyl)amine, pregnan-3β-ol-20-(boc-aminobutyl)amine, pregnan-3β-ol-20-(aminoethyl)amine, pregnan-3β-ol-20-(aminopropyl)amine, pregnan-3β-ol-20-(aminobutyl)amine, O-t-butyldimethylsilylpregnanolone, t-Butyldimethysilylpregnane-3,20-diol, pregnane-3,20-diol, pregnanolone, O-t-butyldimehylsilyl-21-bromopregnanolone, N,N-dimethyl-21-aminopregnanolone, 21-piperidinopregnanolone, N-methyl-21-aminopregnanolone, 21-piperazinopregnanolone, aminothiazole, 3β-hydroxy-20-(pyrid-2-yl)-5α-pregnane, 3β-hydroxy-20-(thiazol-2-yl)-5α-pregnane, 3β-hydroxy-20-(piperidin-2-yl)-5α-pregnane, 3β-hydroxy-20-(thiazol-4-yl)-5α-pregnane, 3β-hydroxy-20-(imidazol-4-yl)-5α-pregnane, 3-methoxymethyltomatidine, 26-amino-3β-hydroxy-16β-methoxy-21-methyl-5α-cholestano-22,26-piperidine, (25S)-26-Acetylamino-3β,16β-dihydroxy-5a-cholestane, 3β-hydroxy-20-(2-aminothiazol-4-yl)-5α-pregnane hydrochloride or a salt thereof. 43. The method of 44. (canceled) 45. The method of 46. (canceled) 47. The method of 48. (canceled) 49. The method of 50. The method of 51. (canceled) 52. The method of 53. (canceled) 54. (canceled) 55. A composition comprising the compound as defined in (i) an antibiotic; (ii) an antiseptic; (iii) a disinfectant; (iv) a diluent; (v) an excipient; (vi) a pharmaceutically acceptable carrier; or (vii) any combination of (i)-(vi). 56. (canceled) 57. The composition of 58. (canceled) 59. A kit comprising at least one of the compounds as defined in 60. (canceled) 61. (canceled) 62. (canceled)CROSS REFERENCE TO RELATED APPLICATIONS
FIELD OF THE INVENTION
REFERENCE TO SEQUENCE LISTING
BACKGROUND OF THE INVENTION
Staphylococci
Bacterial Small-Colony Variants
Cystic Fibrosis
Burn Patients
Bovine Mastitis
Infections Caused by Antibiotic-Resistant Bacteria
Foodborne Bacteria and Illnesses
SUMMARY OF THE INVENTION
Compounds and Compounds for Use
R1 is H; and R2 is —OH or —NR5R6; or
R1 is —OH or —NR5R6; and R2 is H;
R3 is —C(CH3)(NHR9),
R4 is H,
R3 and R4 form together with the carbon atoms to which they are attached form a spirocyclic structure of formula II:
with the proviso that when R1 is NH2 and R2 is H; or R1 is H and R2 is NH2, R10 is not H, or a stereoisomer or mixture of stereoisomers or a salt thereof,
(A) in combination with an aminoglycoside antibiotic for preventing or treating a bacterial infection in a subject; or (B) (a) for preventing or treating an infection caused by an electron transport-deficient bacteria in a subject; or (b) for the disinfection, sterilization and/or antisepsis of an object contaminated with an electron transport-deficient bacteria.
R1 is H; and R2 is —OH or —NR5R6; or
R1 is —OH or —NR5R6; and R2 is H;
R3 is —C(CH3)(NHR9),
R4 is H,
R3 and R4 form together with the carbon atoms to which they are attached form a spirocyclic structure of formula II:
with the proviso that when R3 and R4 form together a spirocyclic structure of formula II, neither R1 nor R2 is —OH; and
with the proviso that when R1 is NH2 and R2 is H; or R1 is H and R2 is NH2, R10 is not H, or a stereoisomer or mixture of stereoisomers or a salt thereof,
(A) in combination with an aminoglycoside antibiotic for preventing or treating a bacterial infection in a subject; or (B) (a) for preventing or treating an infection caused by an electron transport-deficient bacteria in a subject; or (b) for the disinfection, sterilization and/or antisepsis of an object contaminated with an electron transport-deficient bacteria.
iii)
Screening Methods
BRIEF DESCRIPTION OF THE DRAWINGS
DESCRIPTION OF ILLUSTRATIVE EMBODIMENTS
Antimicrobial Activity of Compounds of the Present Invention
Potentiating Activity of Compounds of the Present Invention
DEFINITIONS
Compound
Aliphatic Glycine, Alanine, Valine, Leucine, Isoleucine Hydroxyl or Sulfur/ Serine, Cysteine, Selenocysteine, Selenium-containing Threonine, Methionine Cyclic Proline Aromatic Phenylalanine, Tyrosine, Tryptophan Basic Histidine, Lysine, Arginine Acidic and their Amide Aspartate, Glutamate, Asparagine, Glutamine Microbial Targets
Subjects and Objects
Excipients/Carriers
Routes of Administration
Formulations
Oral
Nasal
Transmucosal or Transdermal (Topical)
Parenteral
Compounds
Protective Group
Salts, Esters, Hydrates and Solvates
Salts
Esters
Hydrates
Prodrugs and Solvates
Stereoisomers, Diastereomers, Enantiomers, Racemates, Tautomers
Dosages
Toxicity and Therapeutic Efficacy
Kits
Example 1
Chemistry
Example 2
General Procedure for Synthesis of Boc-Diaminoalkanes (A)
Example 3
General Procedure for Reductive Amination of 3β-Acetoxy-5-Alpha-Pregnan-20-One (B)
Example 4
General Method for Acidic Deprotections (D)
Example 5
Intermediate N-Formyl Tomatidine (4)
Example 6
N-formyl-3-oxotomatidine (9)
Example 7
Intermediate N-Formyl-3-aminotomatidine (11)
Example 8
N-Boc-1,2-diaminoethane (38)
Example 9
Intermediate N-Formyl-3-(N-Boc-aminoethyl)aminotomatidine (13)
Example 10
3-(N-aminoethyl)-aminotomatidine hydrochloride (14)
Example 11
N-Boc-1,4-diaminobutane (40)
Example 12
Intermediate N-Formyl-3-(N-Boc-aminobutyl)aminotomatidine (15)
Example 13
3-(N-aminobutyl)-aminotomatidine hydrochloride (16)
Example 14
Intermediate N-Formyl-3-(aminohexyl)aminotomatidine (17)
Example 15
3-(N-aminohexyl)-aminotomatidine hydrochloride (18)
Example 16
Intermediate 3β-Acetoxy-20-butylamino-5α-pregnane (47)
Example 17
3β-Hydroxy-20-butylamino-5α-pregnane hydrochloride (48)
Example 18
Intermediate 3β-Acetoxy-20-isopentylamino-5α-pregnane (49)
Example 19
3β-Hydroxy-20-isopentylamino-5α-pregnane hydrochloride (50)
Example 20
Antibacterial Activity of Tomatidine and Derivatives Alone Against a Normal
Biological activities of tomatidine analogs against phenotypes alone or in combination with an aminoglycoside. Combination MIC MIC vs vs ATCC29213 MIC vs ATCC29213 (potentiaton fold with SCVc Compound (μg/ml)a gentamicin)b (μg/ml)a 3 >64 0.06 (8×) High 0.06 High 4 >64 0.25 (2×) Low >8 Low 14 8-16 Active 0.06-0.12 (4-8×) Moderate-high 0.5-1 Moderate 16 16 Active 0.06-0.12 (4-8×) Moderate-high 0.5 Moderate 18 32 0.12-0.25 (2-4×) Moderate 1 Moderate 48 64 0.06 (8×) High 4-8 Moderate 50 >64 0.12 (4×) Moderate >8 Low Compounds 14 to 18, 48 and 50 are racemic mixtures. aCompound tested alone; bThe potentiation fold of compounds is defined as the ratio of the MIC of gentamicin alone and the MIC of gentamicin in combination with the compound used at 8 μg/mL (except for compounds 14 and 16 that were used at 4 μg/mL); and cActivity of the compounds against the SCV strain NewbouldΔhemB (MIC = ≧8 μg/mL: low activity, MIC = 0.5-4 μg/mL: moderate activity, MIC = 0.06-0.25 μg/mL: high activity). Biological activities of Compound 14 against normal phenotype (ATCC 29213) in combination with an aminoglycoside (tobramycin, streptomycin, kanamycin or amikacin). MIC of aminoglycoside (μg/mL) against In combination with Potentiation Aminoglycoside Compound 14 used fold with alone at 4 μg/mL Compound 14a Tobramycin 1 0.125 8 Streptomycin 4-8 1 4-8 Kanamycin 4 0.25 16 Amikacin 4 0.25 16 aThe potentiation fold is defined as the ratio of the MIC of the aminoglycoside alone and the MIC of the aminoglycoside in combination with Compound 14 used at 4 μg/mL. Example 21
Antibacterial Effect of Compound 14 on Normal Antibiotic-Resistant
Antibacterial effect of Compound 14 on normal antibiotic-resistant (MRSA) when used alone or in combination with gentamicin or with kanamycin and cefalexin. Potentiation Potentiation MIC (μg/ml) fold MIC (μg/ml) fold a b c d e f g h i Strain Cpd 14 GEN GEN + GEN + CEF KAN CEF + CEF + CEF + Cpd 14 at 4 Cpd 14 at 4 KAN KAN KAN μg/ml μg/ml (3:2) (3:2) + Cpd (3:2) + Cpd 14 at 4 14 at 4 μg/ml μg/ml MRSA 16 0.5 0.12 4 256 256 128:86 4:3 32 USA100 MRSA 16 0.5 0.06 8 256 >1024 256:171 8:6 32 USA300 MRSA 16 0.25 0.06 4 256 4 4:3 0.5:0.35 8 COL a: MIC of Compound 14 (Cpd 14) against the tested methicillin-resistant b: MIC of gentamicin (GEN) against the tested MRSA strains; c: MIC of gentamicin (GEN) against the tested MRSA strains when Cpd 14 is present at 4 μg/ml; d: The potentiation fold is defined as the ratio of the MIC of gentamicin (GEN) alone and the MIC of GEN in combination with Compound 14 (Cpd 14) used at 4 μg/mL; e: MIC of cefalexin (CEF) against the tested MRSA strains; f: MIC of kanamycin (KAN) against the tested MRSA strains; g: MICs of cefalexin (CEF) and kanamycin (KAN) when used in combination at a ratio of 3:2; h: MICs of cefalexin (CEF) and kanamycin (KAN) when used in combination at a ratio of 3:2, against the tested MRSA strains when Cpd 14 is present at 4 μg/ml; i: The potentiation fold is defined as the ratio of the respective MICs of cefalexin (CEF) and kanamycin (KAN) when used in combination at a ratio of 3:2 and the MICs of CEF and KAN when used in combination at a ratio of 3:2 in the presence of Compound 14 (Cpd 14) used at 4 μg/mL Example 22
Bactericidal Activity of Tomatidine and Compound 14 Alone or in Combination with an Aminoglycoside Against
Example 23
Bactericidal Activity of Tomatidine and Compound 14 Against Small-Colony Variants (SCVs) of Bacterial Species of the Firmicutes Phylum
Example 24
Bactericidal Activity of Tomatidine and Compound 14 Alone or in Combination with an Aminoglycoside Against
Example 25
Testing the Biological Activity of the Compounds of the Invention
Example 26
Inhibitory Effect of Compounds of the Present Invention Measured in Cell Cultures
Example 27
Compound 14 Keeps Some of its Antibacterial Activity Against Tomatidine-Resistant
Antibacterial activity of gentamicin, tomatidine and Compound 14 against (SCV) and tomatidine-resistant SCV mutants (SaR1 and SaR2) Fold Fold increase in increase in Compound MIC (μg/mL) Tomatidine 14 SCV Strain Gentamicin Tomatidine Compound 14 resistanced resistancee 4-8 0.0625 0.5 — — 4-8 >32 8 >512 16 16 >32 4 >512 8 a b c dFold increase in tomatidine resistance as measured by the ratio of the MIC of tomatidine for the indicated mutant and the MIC of tomatidine for the susceptible strain NewbouldΔhemB; and eFold increase in Compound 14 resistance as measured by the ratio of the MIC of Compound 14 for the indicated mutant and the MIC of Compound 14 for the susceptible strain NewbouldΔhemB. Example 28
Generation of
Example 29
Inhibition of ATP Synthase Activity by Compound 14
Example 30
Inhibitory Effect of Compounds of the Present Invention, Alone or in Combination with at Least One Aminoglycoside, Measured During Infection in Animals (In Vivo)
Septicemia Model
Neutropenic Mouse Thigh Model
Lung Infection (Pneumonia) Model
Burned Tissue Model
Mouse Mastitis Model
REFERENCES