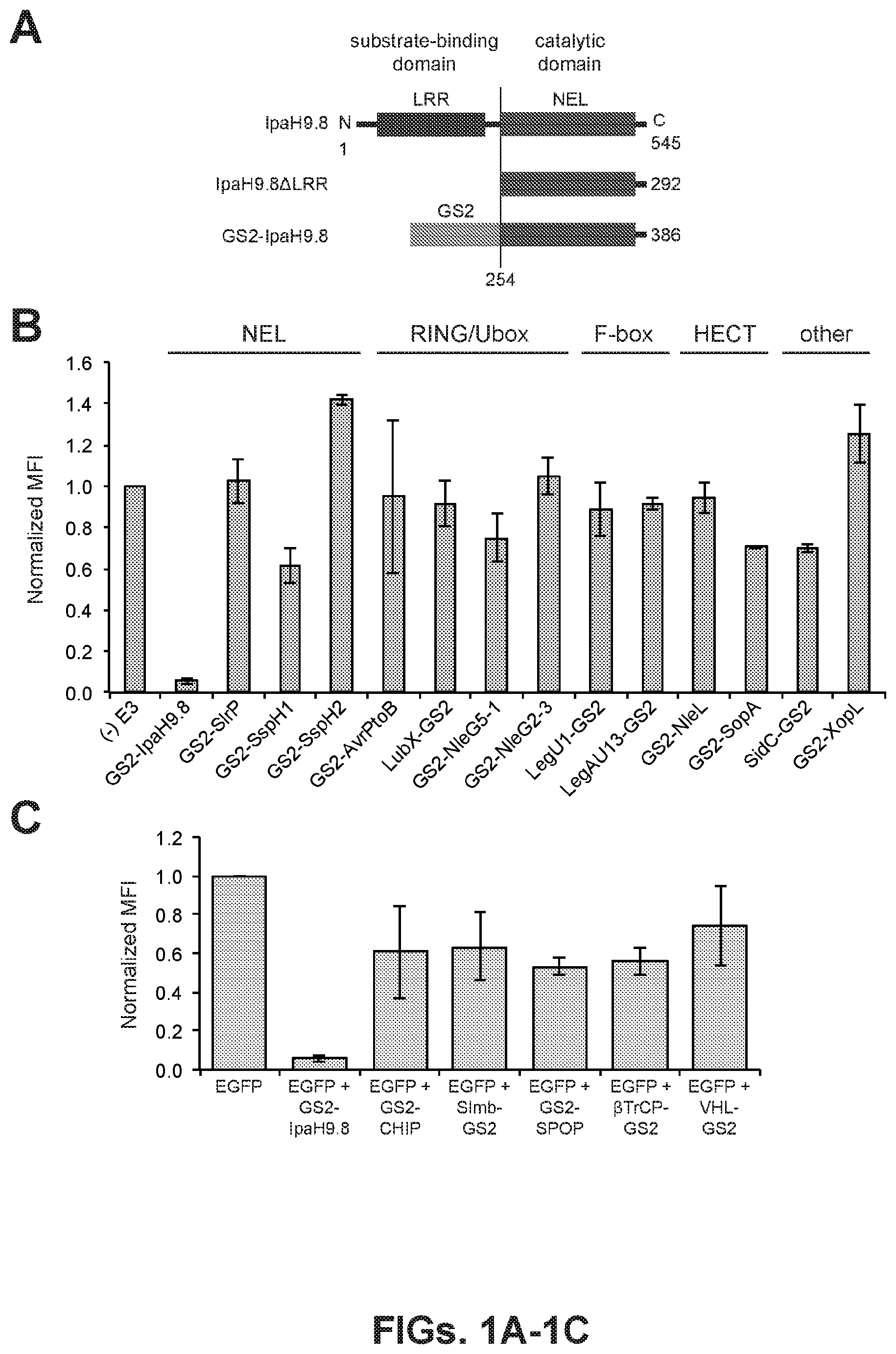
BROAD-SPECTRUM PROTEOME EDITING WITH AN ENGINEERED BACTERIAL UBIQUITIN LIGASE MIMIC
This application claims the priority benefit of U.S. Provisional Patent Application Ser. No. 62/644,055 filed Mar. 16, 2018, which is hereby incorporated by reference in its entirety. The present application relates generally to broad-spectrum proteome editing with an engineered bacterial ubiquitin ligase mimic. Protein function has traditionally been investigated by disrupting the expression of a target gene encoding a protein and analyzing the resulting phenotypic consequences. Such loss-of-function experiments are now routinely performed using gene silencing and genome editing techniques such as antisense oligonucleotides (“ASOs”), RNA interference (“RNAi”), zinc finger nucleases (“ZFNs”), transcription activator-like effector nucleases (“TALENs”), and clustered, regularly interspaced, short palindromic repeat (“CRISPR”)-Cas systems. McManus et al., “Gene Silencing in Mammals by Small Interfering RNAs,” Proteome editing technology represents an orthogonal approach for studying protein function that operates at the post-translational level and has the potential to dissect complicated protein functions at higher resolution than methods targeting DNA or RNA and with post-translational precision. One of the most notable methods involves “inhibition-by-degradation” whereby the machinery of the cellular ubiquitin-proteasome pathway (“UPP”) is hijacked to specifically degrade proteins of interest. The canonical ubiquitination cascade requires the activities of three enzymes—ubiquitin activating enzyme (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3)—which act sequentially to tag proteins for degradation through the covalent attachment of a poly-ubiquitin chain to lysine residues in an energy-dependent manner. E3s are the most heterogeneous class of enzymes in the UPP (there are >600 E3s in humans) and can be classified as HECT (homologous to E6AP C-terminus), RING (really interesting new gene), and RBR (RING-between-RING) depending on the presence of characteristic domains and on the mechanism of ubiquitin transfer to the substrate protein. Buetow et al., “Structural Insights into the Catalysis and Regulation of E3 Ubiquitin Ligases,” To circumvent these issues, protein-based chimeras have been developed where E3 ubiquitin ligases are genetically fused to a protein that binds the target of interest. Following ectopic expression in cells, the engineered protein chimera recruits the E3 to the target protein, leading to its polyubiquitination and subsequent degradation by the proteasome. In the earliest example, protein knockout was achieved by creating an F-box chimera in which β-TrCP was fused to a peptide derived from the E7 protein encoded by human papillomavirus type 16 that is known to interact with retinoblastoma protein pRB (Zhou et al., “Harnessing the Ubiquitination Machinery to Target the Degradation of Specific Cellular Proteins,” More recently, it was shown that a universal proteome editing technology could be extended beyond naturally occurring binding pairs. This approach involved fusing an E3 to a synthetic binding protein such as a single-chain antibody fragment (“scFv”), a designed ankyrin repeat protein (“DARPin”), or a fibronectin type III (“FN3”) monobody. Portnoff et al., “Ubiquibodies, Synthetic E3 Ubiquitin Ligases Endowed With Unnatural Substrate Specificity for Targeted Protein Silencing,” The present application unites expertise in protein-based chimeras whereby novel E3 ubiquitin ligase motifs are genetically fused to a protein that binds the target of interest to address the above challenges and overcome these and other deficiencies in the art. A first aspect of the present application relates to an isolated chimeric molecule. The isolated chimeric molecule comprises a degradation domain comprising an E3 ubiquitin ligase (E3) motif; a targeting domain capable of specifically directing the degradation domain to a substrate, wherein the targeting domain is heterologous to the degradation domain; and a linker coupling the degradation domain to the targeting domain. A second aspect of the present application relates to a method of forming a ribonucleoprotein. The method includes providing a mRNA encoding the isolated chimeric molecule described herein; providing one or more polyadenosine binding proteins (“PABP”); and assembling a ribonucleoprotein complex from the mRNA and the one or more PABPs. A third aspect of the present application relates to a composition comprising the chimeric molecule described herein and a pharmaceutically-acceptable carrier. A fourth aspect of the present application relates to a method of treating a disease. The method includes selecting a subject having a disease and administering the composition described herein to the subject to give the subject an increased expression level of the substrate compared to a subject not afflicted with the disease. A fifth aspect of the present application relates to a method for substrate silencing. The method includes selecting a substrate to be silenced; providing the chimeric molecule described herein; and contacting the substrate with the chimeric molecule under conditions effective to permit the formation of a substrate-molecule complex, wherein the complex mediates the degradation of the substrate to be silenced. A sixth aspect of the present application relates to a method of screening agents for therapeutic efficacy against a disease. The method includes providing a biomolecule whose presence mediates a disease state; providing a test agent comprising (i) a degradation domain comprising an E3 ubiquitin ligase (E3) motif, (ii) a targeting domain capable of specifically directing the degradation domain to the biomolecule, wherein the targeting domain is heterologous to the degradation domain, and (iii) a linker coupling the degradation domain to the targeting domain; contacting the biomolecule with the test agent under conditions effective for the test agent to facilitate degradation of the biomolecule; determining the level of the biomolecule as a result of the contacting; and identifying the test agent which, based on the determining, decreases the level of the biomolecule as being a candidate for therapeutic efficacy against the disease. A seventh aspect of the present application relates to a method of screening for disease biomarkers. The method includes providing a sample of diseased cells expressing one or more ligands; providing a plurality of chimeric molecules comprising (i) a degradation domain comprising an E3 ubiquitin ligase (E3) motif, (ii) a targeting domain capable of specifically directing the degradation domain to the one or more ligands, wherein the targeting domain is heterologous to the degradation domain, and (iii) a linker coupling the degradation domain to the targeting domain; contacting the sample with the plurality of chimeric molecules under conditions effective for the diseased cells to fail to proliferate in the absence of the chimeric molecule; determining which of the chimeric molecules permit the diseased cells to proliferate; and identifying, as biomarkers for the disease, based on the determining the ligands which bind to the chimeric molecules and permit diseased cells to proliferate. Manipulation of the ubiquitin-proteasome pathway to achieve targeted silencing of cellular proteins has emerged as a reliable and customizable strategy for remodeling the mammalian proteome. One such approach involves engineering bifunctional proteins called ubiquibodies that are comprised of a synthetic binding protein fused to an E3 ubiquitin ligase, thus enabling post-translational ubiquitination and degradation of a target protein independent of its function. Here, a panel of new ubiquibodies was designed based on E3 ubiquitin ligase mimics from bacterial pathogens that are capable of effectively interfacing with the mammalian proteasomal degradation machinery for selective removal of proteins of interest. One of these, the The present application thus relates to chimeric molecules, compositions, treatments, pharmaceutical compositions, protein silencing techniques, the elucidation of therapeutic agents, and target screening technologies based on a novel class of chimeric molecules. Such chimeras, termed “ubiquibodies” herein, import the ligase function of an E3 ubiquitin enzyme to generate a molecule possessing target specificity. Such engineered chimeras facilitate the redirection and proteolytic degradation of specific substrate targets, which may not otherwise be bound for the proteasome. In this respect, the targeted elimination of such specific substrates, e.g., intracellular proteins, ascribes a broad range of scientific and clinical indications to the chimeric molecules, compositions, treatments, pharmaceutical compositions, protein silencing techniques, elucidation of therapeutic agents, and screening technologies provided herein. The present application therefore imparts a variety of valuable tools for employing and developing specific prognostic and therapeutic applications based on the proteolytic degradation of aberrantly expressed genes via ubiquitination. Here, the range of E3s that can be functionally reprogrammed as bifunctional uAb chimeras was sought to be broadened. However, in a notable departure from previous efforts involving mammalian E3s, the focus was instead on a set of effector proteins from microbial pathogens that mimic host E3 ubiquitin ligases and hijack the UPP machinery to dampen the innate immune response during infection. Maculins et al., “Bacteria-Host Relationship: Ubiquitin Ligases as Weapons of Invasion. As was noted previously, a major obstacle for the therapeutic development of uAbs is intracellular delivery. Osherovich, L., “Degradation From Within,” It is to be appreciated that certain aspects, modes, embodiments, variations and features of the present application are described below in various levels of detail in order to provide a substantial understanding of the present technology. The definitions of certain terms as used in this specification are provided below. Unless defined otherwise, all technical and scientific terms used herein generally have the same meaning as commonly understood by one of ordinary skill in the art to which the present application belongs. In practicing the subject matter of the present application, many conventional techniques in molecular biology, protein biochemistry, cell biology, immunology, microbiology and recombinant DNA are used. These techniques are well-known and are explained in, e.g., A first aspect of the present application relates to an isolated chimeric molecule. The isolated chimeric molecule comprises a degradation domain comprising an E3 ubiquitin ligase (E3) motif; a targeting domain capable of specifically directing the degradation domain to a substrate, wherein the targeting domain is heterologous to the degradation domain; and a linker coupling the degradation domain to the targeting domain. As used herein, the terms “chimeric molecule”, “chimeric polypeptide”, and “chimeric protein” encompass a molecule having a sequence that includes at least a portion of a full-length sequence of first protein or polypeptide sequence and at least a portion of a full-length sequence of a second protein or polypeptide sequence, where the first and second proteins or polypeptides are different proteins or polypeptides. A chimeric molecule also encompasses proteins or polypeptides that include two or more non-contiguous portions derived from the same protein or polypeptide. A chimeric molecule also encompasses proteins or polypeptides having at least one substitution, wherein the chimeric molecule includes a first protein or polypeptide sequence in which a portion of the first protein or polypeptide sequence has been substituted by a portion of a second protein or polypeptide sequence. As used herein, the term “chimeric molecule” further refers to a molecule possessing a degradation domain and a targeting region, as exemplified herein. The degradation domain and targeting region may be attached in manner known in the art. For example, they may be linked via linker molecule as exemplified herein, fused, covalently attached, non-covalently attached, etc. Moreover, the degradation domain and a targeting region may not be directly attached and/or the attachment may be transient, e.g., if a linker is used, the linker may be cleavable or non-cleavable. As used herein, the term “ubiquitination” refers to the attachment of the protein ubiquitin to lysine residues of other molecules. Ubiquitination of a molecule, such as a peptide or protein, can act as a signal for its rapid cellular degradation, and for targeting to the proteasome complex. As used herein, the terms “ubiquibodies” and “chimeric molecules” are used interchangeably and refer to molecules with at least a degradation domain and a target region, linked by a linker region, as exemplified herein. As used herein the terms “target domain” or “targeting domain” or “targeting moiety” means a polypeptide region bound covalently or non-covalently to a second region within a chimeric molecule, which enhances the concentration of the chimeric molecule or composition in a target sub-cellular location, cell, or tissue relative, as compared to the surrounding locations, cells, and/or tissue. In accord, the chimeric molecules of the present application possess novel E3 ligase motif (referenced herein, for example, as “E3 ligase (EL)”) ubiquitin regions attached to targeting domains, which are accessible for substrate binding. In some embodiments, the substrate is an intracellular substrate. In order to facilitate versatility, however, the targeting domain is derived from a monobody (for example, fibronectin type III domain (“FN3”)), antibody, polyclonal antibody, monoclonal antibody, recombinant antibody, antibody fragment, Fab′, F(ab′)2, Fv, scFv, tascFvs, bis-scFvs, sdAb, VH, VL, Vnar, scFvD10, scFv13R4, scFvD10, humanized antibody, chimeric antibody, complementary determining region (CDR), IgA antibody, IgD antibody, IgE antibody, IgG antibody, IgM antibody, nanobody, intrabody, unibody, minibody, PROTACs, aptameric domains, a ubiquitin binding domain sequence, an E3 binding domain, a non-antibody protein scaffold, Adnectin, Affibody and their two-helix variants, Anticalin, camelid antibody (for example, VHH), knottin, DARPin, and/or Sso7d. The skilled artisan will readily appreciate that such targeting domains, in some embodiments, possess cell/tissue specificity in accord with the novel E3 ligase motif regions described herein. In one embodiment, the targeting domain binds to a non-native substrate. As used herein the term monobody may include any binding portion of an non-immunoglobulin molecule including, for example, FN3 and DARPins, or a polypeptide that contains a binding site, which specifically binds to, or reacts with, a substrate and the like. Monobodies in accordance with the present application include synthetic binding proteins that are constructed using a fibronectin type III domain (“FN3”) as a molecular scaffold. Monobodies are a simple and robust alternative to antibodies for creating target-binding proteins. Monobodies belong to a class of molecules collectively called antibody mimics (or antibody mimetics) and alternative scaffolds that aim to overcome shortcomings of natural antibody molecules. A major advantage of monobodies over conventional antibodies is that monobodies can readily be used as genetically encoded intracellular inhibitors, that is a monobody inhibitor may be expressed in a cell of choice by transfecting the cell with a monobody expression vector. This is attributed to the underlying characteristics of the FN3 scaffold: small (˜90 residues), stable, easy to produce, and its lack of disulfide bonds that makes it possible to produce functional monobodies regardless of the redox potential of the cellular environment, including the reducing environment of the cytoplasm and nucleus. In one embodiment, the targeting domain is a monobody. The monobody may be a fibronectin type III domain (FN3) monobody selected from the group consisting of GS2, Nsa5, and RasInII. The GS2 monobody may, for example, recognize green fluorescent protein (“GFP”). The NSa5 monobody may, for example, be specific for the Src-homology 2 (SH2) domain of SHP2 (Sha et al., “Dissection of the BCR-ABL Signaling Network Using Highly Specific Monobody Inhibitors to the SHP2 SH2 Domains,” A targeting domain that is a monobody may be, for example, a fibronectin type III domain (FN3) monobody. Examples of FN3 monobodies include but are not limited to (with target antigen in parenthesis): GS2 (GFP), Nsa5 (SHP2), RasInI (HRas/KRas), and RasInII (HRas/KRas), 1D10 (CDC34), 1D7 (COPS5), 1C4 (MAP2K5), 2C12 (MAP2K5), 1E2 (SF3A1), 1C2 (USP11), 1A9 (USP11), Ubi4 (ubiquitin), EI1.4.1 (EGFR), EI2.4.6 (EGFR), EI3.4.3 (EGFR), EI4.2.1 (EGFR), EI4.4.2 (EGFR), EI6.2.6 (EGFR), EI6.2.10 (EGFR), E246(EGFR), C743(CEA), IIIa8.2.6 (FcγIIa), IIIa6.2.6 (FcγIIIa), hA2.2.1 (hA33), hA2.2.2 (hA33), hA3.2.1 (hA33), hA3.2.3 (hA33), mA3.2.1 (mA33), mA3.2.2 (mA33), mA3.2.3 (mA33), mA3.2.4 (mA33), mA3.2.5 (mA33), Alb3.2.1 (hAlb), mI2.2.1 (mIgG), HA4 (AblSH2), HA10 (AblSH2), HA16 (AblSH2), HA18 (AblSH2), 159 (vEGFR), MUC16 (MSLN), E2#3 (ERα/EF), E2#4 (ERα/EF), E2#5 (ERα/EF), E2#6 (ERα/EF), E2#7 (ERα/EF), E2#8 (ERα/EF), E2#9 (ERα/EF), E2#10 (ERα/EF), E2#11 (ERα/EF), E2#23 (ERα/EF), E3#2 (ERα/EF), E3#6 (ERα/EF), OHT#31 (ERα/EF), OHT#32 (ERα/EF), OHT#33 (ERα/EF), AB7-A1 (ERα/EF), AB7-B1 (ERα/EF), MBP-74 (MBP), MBP-76 (MBP), MBP-79 (MBP), hSUMO4-33 (hSUMO4), hSUMO-39 (hSUMO4), ySUMO-53 (ySUMO), ySUMO-56 (ySUMO), ySUMO-57 (ySUMO), T14.25 (TNFα), T14.20 (TNFα), FNfn10-3JCL14 (avβ3 integrin), 1C9 (Src SH3), 1F11 (Src SH3), 1F10 (Src SH3), 2G10 (Src SH3), 2B2 (Src SH3), 1E3 (Src SH3), E18 (VEGFR2), E19 (VEGFR2), E26 (VEGFR2), E29 (VEGFR2), FG4.2 (Lysozyme), FG4.1 (Lysozyme), 2L4.1 (Lysozyme), BF4.1 (Lysozyme), BF4.9 (Lysozyme), BF4.4 (Lysozyme), BFs1c4.01 (Lysozyme), BFs1c4.07 (Lysozyme), BFs3_4.02 (Lysozyme), BFs3_4.06 (Lysozyme), BFs3_8.01 (Lysozyme), 10C17C25 (phospho-IκBα), Fn-N22 (SARS N), Fn-N17 (SARS N), FN-N10 (SARS N), gI2.5.3T88I (goat IgG), gI2.5.2 (goat IgG), gI2.5.4 (goat IgG), rI4.5.4 (rabbit IgG), rI4.3.1 (rabbit IgG), rI3.6.6 (rabbit IgG), rI4.3.4 (rabbit IgG), rI3.6.4 (rabbit IgG), and rI4.3.3 (rabbit IgG). As used herein the term antibody may include an immunoglobulin and any antigen-binding portion of an immunoglobulin, e.g., IgG, IgD, IgA, IgM and IgE, or a polypeptide that contains an antigen binding site, which specifically or “immunospecifically binds” to, or “immunoreacts with”, an immunogen, antigen, substrate, and the like. Antibodies can comprise at least one heavy (H) chain and at least one light (L) chain inter-connected by at least one disulfide bond. The term “VH” refers to a heavy chain variable region of an antibody. The term “VL” refers to a light chain variable region of an antibody. In some embodiments, the term “antibody” specifically covers monoclonal and polyclonal antibodies. A “polyclonal antibody” refers to an antibody which has been derived from the sera of animals immunized with an antigen or antigens. A “monoclonal antibody” refers to an antibody produced by a single clone of hybridoma cells. Antibody-related molecules, domains, fragments, portions, etc., useful as targeting domains of the present application include, e.g., but are not limited to, Fab, Fab′ and F(ab′)2, Fd, single-chain Fvs (scFv), single-chain antibodies, disulfide-linked Fvs (sdFv) and fragments comprising either a VLor VHdomain. Examples include: (i) a Fab fragment, a monovalent fragment consisting of the VL, VH, CLand CH1domains; (ii) a F(ab′)2fragment, a bivalent fragment comprising two Fab fragments linked by a disulfide bridge at the hinge region; (iii) a Fd fragment consisting of the VHand CH1domains; (iv) a Fv fragment consisting of the VLand VHdomains of a single arm of an antibody, (v) a dAb fragment (Ward et al., “Binding Activities of a Repertoire of Single Immunoglobulin Variable Domains Secreted From The term “monoclonal antibody” as used herein may include an antibody obtained from a population of substantially homogeneous antibodies, i.e., the individual antibodies comprising the population are identical except for possible naturally occurring mutations that may be present in minor amounts. Nevertheless, the monoclonal antibodies to be used in accordance with the present application may be made by the hybridoma method first described by Kohler et al., “Continuous Cultures of Fused Cells Secreting Antibody of Predefined Specificity,” As used herein, the term “polyclonal antibody” includes, for example, a preparation of antibodies derived from at least two (2) different antibody-producing cell lines. The use of this term includes preparations of at least two (2) antibodies that contain antibodies that specifically bind to different epitopes or regions of an antigen. As used herein, the terms “single chain antibodies” or “single chain Fv (scFv)” may refer to an antibody fusion molecule of the two domains of the Fv fragment, VLand VH. Although the two domains of the Fv fragment, VLand VH, are coded for by separate genes, they can be joined, using recombinant methods, by a synthetic linker that enables them to be made as a single protein chain in which the VLand VHregions pair to form monovalent molecules (known as single chain Fv (scFv). See, e.g., Bird et al., “Single-Chain Antigen-Binding Proteins,” As used herein, the term “variable” may, for example, refer to the fact that certain segments of the variable domains differ extensively in sequence among antibodies. The V domain mediates antigen binding and defines specificity of a particular antibody for its particular antigen. However, the variability is not evenly distributed across the amino acid span of the variable domains. Instead, the V regions consist of relatively invariant stretches called framework regions (FRs) of 15-30 amino acids separated by shorter regions of extreme variability called “hypervariable regions” that are each 9-12 amino acids long. The variable domains of native heavy and light chains each comprise four FRs, largely adopting a β-sheet configuration, connected by three hypervariable regions, which form loops connecting, and in some cases forming part of, the β-sheet structure. The hypervariable regions in each chain are held together in close proximity by the FRs and, with the hypervariable regions from the other chain, contribute to the formation of the antigen-binding site of antibodies. See Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991), which is hereby incorporated by reference in its entirety. The constant domains are not involved directly in binding an antibody to an antigen, but exhibit various effector functions, such as participation of the antibody in antibody dependent cellular cytotoxicity (“ADCC”). The targeting domains of the present application can be, for example, monospecific, bispecific, trispecific or of greater multispecificity. Multispecific targeting domains can be specific for different epitopes of a substrate or can be specific for both a substrate polypeptide of the present application as well as for heterologous compositions, such as a heterologous polypeptide or solid support material. See, e.g., WO 93/17715; WO 92/08802; WO 91/00360; WO 92/05793; Tutt et al., “Trispecific F(ab′)3 Derivatives That Use Cooperative Signaling Via the TCR/CD3 Complex and CD2 to Activate and Redirect Resting Cytotoxic T Cells,” Techniques for generating targeting domains directed to target substrates are well known to those skilled in the art. Examples of such techniques include, but are not limited to, e.g., those involving display libraries, xeno or humab mice, hybridomas, and the like. Target polypeptides—from which a targeting domain is derived—within the scope of the present application include any polypeptide or polypeptide derivative which is capable of exhibiting antigenicity. Examples include, but are not limited to, substrate and fragments thereof. In some embodiments, the targeting domain is a single-chain antibody. Single chain antibodies (“scFv”) are genetically engineered antibodies that consist of the variable domain of a heavy chain at the amino terminus joined to the variable domain of a light chain by a flexible region. In some embodiments, scFv are generated by PCR from hybridoma cell lines that express monoclonal antibodies (mAbs) with known target specificity, or they are selected by phage display from libraries isolated from spleen cells or lymphocytes, and preserve the affinity of the parent antibody. Employing a protocol to identify intracellular substrates, the yeast two-hybrid technology serves to identify candidate scFv—protein interactions. Such a system is useful to predict whether or not a scFv will be able to recognize its target substrate in vivo. See Portner-Taliana et al., “Identification of Protein Single chain Antibody Interactions In Vivo Using Two-hybrid Protocols,” Typically, scFv, hybrid antibodies or hybrid antibody fragments that are cloned into a display vector can be selected against the appropriate antigen in order to identify variants that maintained good binding activity, because the antibody or antibody fragment will be present on the surface of the phage or phagemid particle. See e.g., Barbas III et al., In general, expression vectors useful in recombinant DNA techniques are often in the form of plasmids. However, the present application is intended to include such other forms of expression vectors that are not technically plasmids, such as viral vectors, e.g., replication defective retroviruses, adenoviruses and adeno-associated viruses, which serve equivalent functions. Such viral vectors permit infection of a subject and expression in that subject of a compound. The expression control sequences are typically eukaryotic promoter systems in vectors capable of transforming or transfecting eukaryotic host cells. Once the vector has been incorporated into the appropriate host, the host is maintained under conditions suitable for high level expression of the nucleotide sequences encoding the target domain, and the collection and purification of the substrate binding agent, e.g., cross-reacting anti-substrate antibodies. See, generally, U.S. Patent Publication No. 2002/0199213, which is hereby incorporated by reference in its entirety. Vectors can also encode signal peptide, e.g., pectate lyase, useful to direct the secretion of extracellular antibody fragments. See U.S. Pat. No. 5,576,195, which is hereby incorporated by reference in its entirety. As used herein, the terms “degradation domain” or “degradation region” includes a portion of a chimeric molecule that is capable of facilitating the ubiquitination of a substrate. The degradation domain may have a second “binding” region for interaction with a native binding protein. The binding region can be modified as to possess one or more mutations, substitutions, deletions, or may be deleted entirely. The degradation domain may contain E3 mimics with folds similar to eukaryotic E3s such as HECT-type, RING or U-box (RING/U-box)-type, and F-box domains, as well as unconventional E3s with folds unlike any other eukaryotic E3s such as NEL, XL-box-containing, and SidC. The degradation domain relates to polypeptides or polypeptide regions capable of modifying substrates by attaching one or more ubiquitin molecules and/or ubiquitin-like molecules to the substrates. In this regard, such a region comports with the well-known ubiquitination cascade—involving the coordinated action of the E1, E2, and E3 enzymes—which functions to activate and concomitantly conjugate ubiquitin to a substrate. In some embodiments, the motif is a ubiquitin region composed of a novel E3 ligase, or fragment thereof, which catalyzes the transfer of ubiquitin in a substrate-specific manner. See Qian et al., “Engineering a Ubiquitin Ligase Reveals Conformational Flexibility Required for Ubiquitin Transfer,” As used herein, the terms “modification(s)” or “amino acid modification” of a polypeptide, protein, region, domain, or the like, refers to a change in the native sequence such as a deletion, addition or substation of a desired residue. Such modified polypeptides are prepared by introducing appropriate nucleotide changes into the antibody nucleic acid, or by peptide synthesis. Any combination of deletion, insertion, and substitution is made to obtain the antibody of interest, as long as the obtained antibody possesses the desired properties. The modification also includes the change of the pattern of glycosylation of the protein. A useful method for identification of preferred locations for mutagenesis is called “alanine scanning mutagenesis” as described by Cunningham and Wells, “High-Resolution Epitope Mapping of hGH-Receptor Interactions by Alanine-Scanning Mutagenesis,” The terms “polypeptide,” “protein,” and “peptide” are used herein interchangeably herein to refer to amino acid chains in which the amino acid residues are linked by peptide bonds or modified peptide bonds. The amino acid chains can be of any length of greater than two amino acids. Unless otherwise specified, the terms “polypeptide,” “protein,” and “peptide” also encompass various modified forms thereof. Such modified forms may be naturally occurring modified forms or chemically modified forms. Examples of modified forms include, but are not limited to, glycosylated forms, phosphorylated forms, myristoylated forms, palmitoylated forms, ribosylated forms, acetylated forms, ubiquitinated forms, etc. Modifications also include intra-molecular crosslinking and covalent attachment to various moieties such as lipids, flavin, biotin, polyethylene glycol or derivatives thereof, etc. In addition, modifications may also include cyclization, branching and cross-linking. Further, amino acids other than the conventional twenty amino acids encoded by genes may also be included in a polypeptide. In one embodiment, the E3 ubiquitin ligase motif (E3), also referred to herein as EL or NEL, comprises a modified binding region which inhibits or decreases binding to said substrate compared to said E3 motif without the modified binding region. In another embodiment, the modification is a mutation or deletion in the binding region. As used herein, the terms “variant” or “mutant” are used to refer to a protein or peptide which differs from a naturally occurring protein or peptide, i.e., the “prototype” or “wild-type” protein, by modifications to the naturally occurring protein or peptide, but which maintains the basic protein and side chain structure of the naturally occurring form. Such changes include, but are not limited to: changes in one, few, or even several amino acid side chains; changes in one, few or several amino acids, including deletions, e.g., a truncated version of the protein or peptide, insertions and/or substitutions; changes in stereochemistry of one or a few atoms; and/or minor derivatizations, including but not limited to: methylation, glycosylation, phosphorylation, acetylation, myristoylation, prenylation, palmitation, amidation and/or addition of glycosylphosphatidyl inositol. A “variant” or “mutant” can have enhanced, decreased, changed, or substantially similar properties as compared to the naturally occurring protein or peptide. In some embodiments, the degradation domain of the chimeric molecule lacks an endogenous substrate recognition region, i.e., a portion of the polypeptide that interacts with a natural or native binding partner. The E3 motif of the degradation domain may possess a modified binding domain which inhibits or decreases binding to a substrate compared to the E3 motif without the modified binding region. Nevertheless, the E3 motif permits proteolysis of a substrate in some embodiments. In some embodiments, the modification is a mutation, substitution, or deletion of the binding region. The substitution can be an amino acid substitution such as a conservative or a non-conservative amino acid substitution. Non-conservative amino acid substitutions of the E3 motif, are substitutions in which an alkyl amino acid is substituted for an amino acid other than an alkyl amino acid in the sequence, an aromatic amino acid is substituted for an amino acid other than an aromatic amino acid in the E3 motif, a sulfur-containing amino acid is substituted for an amino acid other than a sulfur-containing amino acid in the E3 motif, a hydroxy-containing amino acid is substituted for an amino acid other than a hydroxy-containing amino acid in the E3 motif, an acidic amino acid is substituted for an amino acid other than an acidic amino acid in the E3 motif, a basic amino acid is substituted for an amino acid other than a basic amino acid in the E3 motif, or a dibasic monocarboxylic amino acid is substituted for an amino acid other than a dibasic monocarboxylic amino acid in the E3 motif. Among the common amino acids, for example, “non-conservative amino acid substitutions” are illustrated by a substitution of an amino acids from one of the following groups with an amino acid that is not from the same group, as follows: (1) glycine, alanine, (2) valine, leucine, and isoleucine, (3) phenylalanine, tyrosine, and tryptophan, (4) cysteine and methionine, (5) serine and threonine, (6) aspartate and glutamate, (7) glutamine and asparagine, and (8) lysine, arginine and histidine. Conservative or non-conservative amino acid changes in, e.g., the E3 motif, can be introduced by substituting appropriate nucleotides for the nucleotides encoding such a region. These modifications can be obtained, for example, by oligonucleotide-directed mutagenesis, linker-scanning mutagenesis, mutagenesis using the polymerase chain reaction, and the like. Ausubel et al. (eds.), Short Protocols in Molecular Biology, 5th Edition, John Wiley & Sons, Inc. (2002); see generally, McPherson (ed.), Directed Mutagenesis: A Practical Approach, IRL Press (1991), which are hereby incorporated by reference in their entirety. A useful method for identification of locations for sequence variation is called “alanine scanning mutagenesis” a described by Cunningham and Wells “Protein Engineering of Antibody Binding Sites: Recovery of Specific Activity in an Anti-Digoxin Single-Chain Fv Analogue Produced in Ubiquitin ligase families include, but are not limited to, the homologous to E6-associated protein C-terminus (“HECT”) domain ligases, which concerns the transfer of ubiquitin from the E2 conjugase to the substrate, the Really Interesting New Gene (“RING”) domain ligases, which bind E2, may mediate enzymatic activity in the E2-E3 complex, and the U-box ubiquitin family of ligases (“UULs”), which constitute a family of modified RING motif ligases without the full complement of Zn2+-binding ligands. See Colas et al., “Targeted Modification and Transportation of Cellular Proteins.” U-box ubiquitin ligases (“ULLs”) are characterized as having a protein domain, the U-box, which is structurally related to the RING finger, typical of many other ubiquitin ligases. In humans, the UUL-encoding genes include, but are not limited to, UBE4A and UBE4B genes (also respectively termed UFD2b and UFD2a), CHIP (also termed STUB1), UIPS (also termed UBOXS), PRP19 (also termed PRPF19 or SNEV), CYC4 (also termed PPIL2 or Cyp-60), WDSUB1, and ACT1 (also termed TRAF3IP2). See Marin, I., “Ancient Origin of Animal U-box Ubiquitin Ligases.” While HECT E3 ligases have a direct role in catalysis during ubiquitination, RING and U-box E3 proteins facilitate protein ubiquitination by acting as adaptor molecules that recruit E2 and substrate molecules to promote substrate ubiquitination. Although many RING-type E3 ligases, such as MDM2 (murine double minute clone 2 oncoprotein) and c-Cbl, may act alone, others are found as components of much larger multi-protein complexes, such as the anaphase-promoting complex (“APC”). Taken together, these multifaceted properties and interactions enable E3 enzymes to provide a powerful, and specific, mechanism for protein clearance within all cells of eukaryotic organisms. Ardley et al., “E3 Ubiquitin Ligases.” Functional information concerning the E3 gene products is variable, nonetheless. The U-box protein CHIP acts both as a co-chaperone, together with chaperones such as, e.g., Hsc70, Hsp70 and Hsp90, and as a ubiquitin ligase, alone or as part of complexes that may include other E3 proteins. See id. The selectivity of the ubiquitin proteasome system for a particular substrate nevertheless relies on the interaction between a ubiquitin-conjugating enzyme, e.g., E2, and a ubiquitin-protein ligase. Post-translational modifications of the protein substrate, such as, e.g., phosphorylation or hydroxylation, are often required prior to ubiquitination. In this way, the precise spatio-temporal targeting and degradation for a particular substrate can be achieved. The E3 motif of the degradation domain disclosed herein possesses a functional E3 ligase that is capable of ubiquitinating a substrate without steric disruption from native binding partners. In addition to the E3 motif, in some embodiments, the degradation domain possesses a ligase that is an E3 mimic with folds similar to eukaryotic E3s such as HECT-type, RING or U-box (RING/U-box)-type, and F-box domains, as well as unconventional E3s with folds unlike any other eukaryotic E3s such as NEL, XL-box-containing, and SidC. Such domains may possess cell or tissue specificity. The E3 motif of the chimeric molecule may, in one embodiment, possess cell-type specific or tissue specific ligase function for, but not limited to, skin cells, muscle cells, epithelial cells, endothelial cells, stem cells, umbilical vessel cells, corneal cells, cardiomyocytes, aortic cells, corneal epithelial cells, somatic cells, fibroblasts, keratinocytes, melanocytes, adipose cells, bone cells, osteoblasts, airway cells, microvascular cells, mammary cells, vascular cells, chondrocytes, placental cells, hepatocytes, glial cells, epidermal cells, limbal stem cells, periodontal stem cells, bone marrow stromal cells, hybridoma cells, kidney cells, pancreatic islets, articular chondrocytes, neuroblasts, lymphocytes, and erythrocytes, and/or any combination thereof. In one embodiment, the degradation domain is from a bacterial pathogen, the pathogen being optionally selected from In one embodiment, the degradation domain is a member of the IpaH family proteins are widely conserved among animal and plant pathogens, including Ubiquitin ligase families also include the “F-box” ligases-as in the Skp1-Cullin1-F-box (“SCF”) protein complex—which binds to a ubiquitinated substrate, such as, e.g., Cdc 4, which subsequently interacts with a target protein, such as, Sic1 or Grr1, which then binds Cln. See Bai et al., “SKP1 Connects Cell Cycle Regulators to the Ubiquitin Proteolysis Machinery through a Novel Motif, the F-Box,” The F-box is a protein motif of approximately 50 amino acids that functions as a site of protein-protein interaction. See, e.g., Kipreos et al., “The F-box Protein family.” The least variant positions within the F-box motif include positions 8 (92% of the 234 F-box proteins used for the consensus have leucine or methionine), 9 (92% proline), 16 (86% isoleucine or valine), 20 (81% leucine or methionine), and 32 (92% serine or cysteine). Id. This lack of a strict consensus guides the skilled artisan to employ multiple search algorithms for detecting F-box sequences. Two algorithms, for example, can be found in the Prosite and Pfam databases. Occasionally, one database will give a significant score to an F-box in a given protein when the other does not detect it, so both databases should be searched. Id. Expression of the chimeric molecules of the present application in prokaryotes is most often carried out in Examples of suitable inducible non-fusion In some embodiments, a nucleic acid encoding a chimeric molecule of the present application—including a degradation domain and a targeting region—is expressed in mammalian cells using a mammalian expression vector. Examples of mammalian expression vectors include, e.g., but are not limited to, pcDNA3, pCDM8 (Seed, “An LFA-3 cDNA Encodes a Phospholipid-Linked Membrane Protein Homologous to its Receptor CD2 Notwithstanding chimeric molecule E3 degradation domains and targeting domain expression, the function of such domains/regions imparts the specificity of the present application. A known or unknown substrate is bound by the targeting domain for subsequent ubiquitination via the degradation domain. In some embodiments, the substrates include, but are not limited to, intracellular substrates, extracellular substrates, modified substrates, glycosylated substrates, farnesylated substrates, post translationally modified substrates, phosphorylated substrates, and other modifications known in the art. In some embodiments, the substrates include, but are not limited to, fluorescent protein, histone protein, nuclear localization signal (NLS), H-Ras protein, Src-homology 2 domain-containing phosphatase 2 (SHP2), β-galactosidase, gpD, Hsp70, MBP, CDC34, COPS5, MAP2K5, SF3A1, USP11, ubiquitin, EGFR, CEA, FcγIIa, FcγIIIa, hA33, mA33, hAlb, mIgG, AblSH2, vEGFR, MSLN, ERα/EF, hSUMO4, ySUMO, TNFα, avβ3 integrin, Src SH3, Lysozyme, phospho-IκBα, SARS N, goat IgG, rabbit IgG, post-translationally modified proteins, fibrillin, huntingtin, tumorigenic proteins, p53, Rb, adhesion proteins, receptors, cell-cycle proteins, checkpoint proteins, HFE, ATP7B, prion proteins, viral proteins, bacterial proteins, parasitic proteins, fungal proteins, DNA binding proteins, metabolic proteins, regulatory proteins, structural proteins, enzymes, immunogenic proteins, autoimmunogenic proteins, immunogens, antigens, pathogenic proteins, and the like. In one embodiment, the substrate is a fluorescent protein, for example, green fluorescent protein, emerald fluorescent protein, venus fluorescent protein, cerulean fluorescent protein, and enhanced cyan fluorescent protein. As used herein, the term “amino acid” includes naturally-occurring amino acids, L-amino acids, D-amino acids, and synthetic amino acids, as well as amino acid analogs and amino acid mimetics that function in a manner similar to the naturally-occurring amino acids. Naturally-occurring amino acids are those encoded by the genetic code, as well as those amino acids that are later modified, e.g., hydroxyproline, γ-carboxyglutamate, and O-phosphoserine. Amino acid analogs refers to compounds that have the same basic chemical structure as a naturally-occurring amino acid, e.g., an a-carbon that is bound to a hydrogen, a carboxyl group, an amino group, and an R group, e.g., homoserine, norleucine, methionine sulfoxide, methionine methyl sulfonium. Such analogs have modified R-groups, e.g., norleucine, or modified peptide backbones, but retain the same basic chemical structure as a naturally-occurring amino acid. Amino acid mimetics refers to chemical compounds that have a structure that is different from the general chemical structure of an amino acid, but that functions in a manner similar to a naturally-occurring amino acid. Amino acids can be referred to herein by either their commonly known three letter symbols or by the one-letter symbols recommended by the IUPAC-IUB Biochemical Nomenclature Commission. An exemplary E3 ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase AvrPtoB, which is a U-box motif, from The E3 ubiquitin ligase AvrPtoB from A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase IpaH0722, which is a novel E3 ligase (also referred to herein as NEL or EL), from The E3 ubiquitin ligase IpaH0722, which is a novel E3 ligase, from A further exemplary E3 ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase IpaH1.4, which is a novel E3 ligase, from The E3 ubiquitin ligase IpaH1.4, which is a novel E3 ligase, from A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase IpaH2.5, which is a novel E3 ligase, from The E3 ubiquitin ligase IpaH2.5, which is a novel E3 ligase, from A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase IpaH4.5, which is a novel E3 ligase, from The E3 ubiquitin ligase IpaH4.5, which is a novel E3 ligase, from A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase IpaH7.8, which is a novel E3 ligase, from The E3 ubiquitin ligase IpaH7.8, which is a novel E3 ubiquitin ligase, from A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase IpaH9.8, which is a novel E3 ligase, from The E3 ubiquitin ligase IpaH9.8, which is a novel E3 ligase, from A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase LegAU13, which is a F-box motif, from The E3 ubiquitin ligase LegAU13, which is a F-box motif, from A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase LegU1, which is a F-box motif, from The E3 ubiquitin ligase LegU1, which is a F-box motif, from A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase LubX, which is a U-box motif, from The E3 ubiquitin ligase LubX, which is a U-box motif, from A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase NleG2-3, which is a U-box motif, Enterohemorrhagic The E3 ubiquitin ligase NleG2-3, which is a U-box motif, from Enterohemorrhagic A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 Ubiquitin Ligase NleG5-1, which is a U-box motif, from Enterohemorrhagic The E3 ubiquitin ligase NleG5-1, which is a U-box motif, from Enterohemorrhagic A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase NleL, which is a HECT motif, from Enterohemorrhagic A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase NleL, which is a HECT motif, from Enterohemorrhagic A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase SidC, which is an unconventional motif, from The E3 ubiquitin ligase SidC, which is an unconventional motif, from A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase SlrP, which is a NEL motif, from EHEC O157:H7, and has the amino acid sequence of SEQ ID NO: 29: The E3 ubiquitin ligase SlrP, which is a NEL motif, from EHEC O157:H7, has the nucleotide sequence of SEQ ID NO: 30 as follows: A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase SopA, which is a HECT motif, from The E3 ubiquitin ligase SopA, which is a HECT motif, from A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase SspH1, which is a novel E3 ligase motif, from The E3 ubiquitin ligase SspH1, which is a novel E3 ligase motif, from A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase SspH2, which is a novel E3 ligase motif, from The E3 ubiquitin ligase SspH2, which is a novel E3 ligase motif, from A further exemplary E3 ubiquitin ligase that is useful as a degradation domain in accordance with the present application includes the E3 ubiquitin ligase XopL, which is an unconventional motif, from The E3 ubiquitin ligase XopL, which is an unconventional motif, from Although targeting domains possess intrinsic binding interactions, e.g., secondary, tertiary or quaternary flexibility, there must still be flexibility with respect to the association with the E3 motif ubiquitin region. In this regard, absence adequate spacing, it is possible for the E3 motif to sterically hinder the substrate-target domain interaction. As such, the present application employs polypeptide linkers of sufficient length to prevent the steric disruption of binding between the targeting domain and the substrate, in some embodiments. In some embodiments, the targeting domain is covalently attached to the ubiquitin region via a linker that may be cleavable or non-cleavable under physiological conditions. The linker can entail an organic moiety comprising a nucleophilic or electrophilic reacting group which allows covalent attachment to the targeting domain to the ubiquitin region agent. In some embodiments, the linker is an enol ether, ketal, imine, oxime, hydrazone, semicarbazone, acylimide, or methylene radical. The linker may be an acid-cleavable linker, a hydrolytically cleavable linker, or enzymatically-cleavable linker, in some embodiments. Peptide-based linking groups are cleaved by enzymes such as peptidases and proteases in cells. Peptide-based cleavable linking groups are peptide bonds formed between amino acids to yield oligopeptides, e.g., dipeptides, tripeptides, and poly-peptides. Peptide-based cleavable groups do not include the amide group (—C(O)NH—). The amide group can be formed between any alkylene, alkenylene or alkynelene. A peptide bond is a special type of amide bond formed between amino acids to yield peptides and proteins. The peptide based cleavage group is generally limited to the peptide bond, i.e., the amide bond, formed between amino acids yielding peptides and proteins and does not include the entire amide functional group. Peptide cleavable linking groups have the general formula —NHCHR1C(O)NHCHR2C(O)—, where R1 and R2 are the R groups of the two adjacent amino acids. These candidates can be evaluated using methods analogous to those described above. For in vitro applications, appropriate linkers, which can be cross-linking agents for use for conjugating a polypeptide to a solid support, include a variety of agents that can react with a functional group present on a surface of the support, or with the polypeptide, or both. Reagents useful as cross-linking agents include homo-bi-functional and, in particular, hetero-bi-functional reagents. Useful bi-functional cross-linking agents include, but are not limited to, N-SIAB, dimaleimide, DTNB, N-SATA, N-SPDP, SMCC and 6-HYNIC. A cross-linking agent can be selected to provide a selectively cleavable bond between a polypeptide and the solid support. For example, a photolabile cross-linker, such as 3-amino-(2-nitrophenyl)propionic acid can be employed as a means for cleaving a polypeptide from a solid support. See Brown et al., “A Single-Bead Decode Strategy Using Electrospray Ionization Mass Spectrometry and a New Photolabile Linker: 3-amino-3-(2-nitrophenyl)propionic Acid,” An antibody, polypeptide, or fragment thereof, such as a targeting domain, can be immobilized on a solid support, such as a bead, through a covalent amide bond formed between a carboxyl group functionalized bead and the amino terminus of the polypeptide or, conversely, through a covalent amide bond formed between an amino group functionalized bead and the carboxyl terminus of the polypeptide. In addition, a bi-functional trityl linker can be attached to the support, e.g, to the 4-nitrophenyl active ester on a resin, such as a Wang resin, through an amino group or a carboxyl group on the resin via an amino resin. Using a bi-functional trityl approach, the solid support can require treatment with a volatile acid, such as formic acid or trifluoracetic acid to ensure that the polypeptide is cleaved and can be removed. In such a case, the polypeptide can be deposited as a beadless patch at the bottom of a well of a solid support or on the flat surface of a solid support. After addition of a matrix solution, the polypeptide can be desorbed into a MS. It will be readily apparent to the skilled artisan that the methods and techniques described above can be employed for the chimeric molecule of the present application, including its constituent parts, e.g., degradation domains, ubiquitin regions, target regions, and modifications thereof, as well as the linker molecules, as described above. A second aspect of the present application relates to a method of forming a ribonucleoprotein. The method includes providing a mRNA encoding the isolated chimeric molecule described herein; providing one or more polyadenosine binding proteins (“PABP”); and assembling a ribonucleoprotein complex from the mRNA and the one or more PABPs. In one embodiment, the mRNA comprises a 3′-terminal polyadenosine (poly A) tail. The chimeric molecule described in this aspect is carried out in accordance with the previously described aspect of the application. The polyadenosine binding proteins (“PABP”) (also referred to as poly(A)-binding proteins) as described herein refer to a RNA-binding protein which binds to the poly(A) tail of mRNA. The poly(A) tail is located on the 3′ end of mRNA and is 200-250 nucleotides long. The binding protein is also involved in mRNA precursors by helping polyadenylate polymerase add the poly(A) nucleotide tail to the pre-mRNA before translation. The nuclear isoform selectively binds to around 50 nucleotides and stimulates the activity of polyadenylate polymerase by increasing its affinity towards RNA. Poly(A)-binding protein is also present during stages of mRNA metabolism including nonsense-mediated decay and nucleocytoplasmic trafficking. The poly(A)-binding protein may also protect the tail from degradation and regulate mRNA production. The ribonucleoprotein, which is also referred to herein as a nanoplex, may, in one embodiment, be a nanoparticle. In a preferred embodiment, the nanoplex includes a nanoparticle. The ribonucleoprotein or nanoplex is a complex formed by a drug nanoparticle with an oppositely charged polyelectrolyte. Both cationic and anionic drugs form complexes with oppositely charged polyelectrolytes. Compared with other nanostructures, the yield of Nanoplex is generally greater and the complexation efficiency is generally better. Nanoplexes are also easier to prepare as compared to other nanostructures. Ribonucleoprotein or nanoplex formulation according to the present application is characterized through the production yield, complexation efficiency, drug loading, particle size and zeta potential using scanning electron microscopy, differential scanning calorimetry, X-ray diffraction and dialysis studies. Nanoplexes have wide-ranging applications in different fields such as cancer therapy, gene drug delivery, drug delivery to the brain and protein and peptide drug delivery. The ribonucleoprotein or nanoplex can have any suitable size. In at least one embodiment, ribonucleoprotein or nanoplex is less than about 200 nm in diameter, less than about 100 nm in diameter, less than about 95 nm, less than about 90 nm, less than about 85 nm, less than about 80 nm, less than about 75 nm, less than about 70 nm, less than about 65 nm, less than about 60 nm, less than about 55 nm, less than about 50 nm, less than about 45 nm, less than about 40 nm, less than about 35 nm, less than about 30 nm, less than about 25 nm, less than about 20 nm, less than about 15 nm, less than about 10 nm, less than about 9 nm, less than about 8 nm, less than about 7 nm, less than about 6 nm, less than about 5 nm, less than about 4 nm, less than about 3 nm, less than about 2 nm, or less than about 1 nm. Nanoparticles having a diameter in a range having an upper limit of about 100 nm, about 95 nm, about 90 nm, about 85 nm, about 80 nm, about 75 nm, about 70 nm, about 65 nm, about 60 nm, about 55 nm, about 50 nm, about 45 nm, about 40 nm, about 35 nm, about 30 nm, about 25 nm, about 20 nm, about 15 nm, about 10 nm, about 9 nm, about 8 nm, about 7 nm, about 6 nm, about 5 nm, about 4 nm, about 3 nm, or about 2 nm and a lower limit of about 95 nm, about 90 nm, about 85 nm, about 80 nm, about 75 nm, about 70 nm, about 65 nm, about 60 nm, about 55 nm, about 50 nm, about 45 nm, about 40 nm, about 35 nm, about 30 nm, about 25 nm, about 20 nm, about 15 nm, about 10 nm, about 9 nm, about 8 nm, about 7 nm, about 6 nm, about 5 nm, about 4 nm, about 3 nm, about 2 nm, or about 1 nm, and any combination thereof, are also contemplated. The present application further provides that in certain embodiments the nanoplex ranges in size from about 50 nm to about 200 nm or from about 10 nm to about 100 nm. In certain embodiments, the size of the nanoplex is about 50 nm to about 150 nm. In other embodiments, the size of the nanoplex can be about 50 nm, about 75 nm, or about 100 nm. The size of the ribonucleoprotein and/or nanoplex may be optimized to localize at a particular location within a subject. For example, the ribonucleoprotein and/or nanoplex may be optimized so that it can move into various organs of a subject. In one embodiment, the ribonucleoprotein and/or nanoplex contains an organic gel. The ribonucleoprotein and/or nanoplex of the present application can have any suitable shape. For example, the present ribonucleoprotein and/or nanoplex can have a shape of a mesh, sphere, square, rectangle, triangle, circular disc, cube-like shape, cube, rectangular parallelepiped (cuboid), cone, cylinder, prism, polyhedral, pyramid, right-angled circular cylinder, rod, branched cylindrical, and other regular or irregular shape. The polymer matrix of the ribonucleoprotein and/or nanoplex may, in one embodiment, contain entangled and covalently bound polymers. In one embodiment, the matrix is a hydrogel. In one embodiment, the number of polymeric units in the ribonucleoprotein and/or nanoplex matrix ranges from 10 to 5000, for instance from 20 to 400, for each particle formed from the polymeric units. In another embodiment, the number of polymeric units ranges from 10,000 to 200,000, for instance from 15,000 to 200,000 polymeric units. The ribonucleoprotein and/or nanoplex according to the present application may include a functionalized surface. In one embodiment, the ribonucleoprotein and/or nanoplex is negatively functionalized. Alternatively, the ribonucleoprotein and/or nanoplex may be positively functionalized. In other embodiments, the ribonucleoprotein and/or nanoplex has a no charge or is neutral. In one embodiment, the ribonucleoprotein and/or nanoplex is biodegradable. In another embodiment, the ribonucleoprotein and/or nanoplex is non-toxic. In one embodiment, the ribonucleoprotein and/or nanoplex may further include at least one stabilizer. The stabilizer may be adsorbed on the surfaces of the ribonucleoprotein and/or nanoplex. The ribonucleoprotein and/or nanoplex may be dispersed into a liquid medium, and the stabilizer may be employed as an adjuvant to aid in the separation of the individual ribonucleoprotein and/or nanoplex during a dispersion process. The ability of a stabilizer to aid in the separation of the individual ribonucleoprotein and/or nanoplex may be determined by comparing the dispersion processes for a composition containing the stabilizer and a control composition without the stabilizer. The ability of a stabilizer to aid in the separation of individual nanoparticles may be indicated by shorter dispersion times. Alternatively, the stabilizer may be employed to promote stability of the dispersed nanoplex in the liquid medium, preferably an aqueous medium. A third aspect of the present application relates to a composition comprising the chimeric molecule described herein and a pharmaceutically-acceptable carrier. According to the methods of the present application, the chimeric molecule can be incorporated into pharmaceutical compositions suitable for administration. As used herein, the term “pharmaceutically-acceptable carrier” is intended to include any and all solvents, dispersion media, coatings, antibacterial and antifungal compounds, isotonic and absorption delaying compounds, and the like, compatible with pharmaceutical administration. The chimeric molecule described in this aspect is carried out in accordance with the previously described aspects of the present application. The pharmaceutical compositions generally entail recombinant or substantially purified chimeric molecules and a pharmaceutically-acceptable carrier in a form suitable for administration to a subject. Pharmaceutically-acceptable carriers are determined in part by the particular composition being administered, as well as by the particular method used to administer the composition. Accordingly, there is a wide variety of suitable formulations of pharmaceutical compositions for administering the protein compositions. See, e.g., As used herein, the term pharmaceutically-acceptable carrier includes, for example, a non-toxic, inert solid, semi-solid or liquid filler, diluent, encapsulating material or formulation auxiliary of any type. The compounds of the present application can be administered orally, parenterally, for example, subcutaneously, intravenously, intramuscularly, intraperitoneally, by intranasal instillation, or by application to mucous membranes, such as, that of the nose, throat, and bronchial tubes. They may be administered alone or with suitable pharmaceutical carriers, and can be in solid or liquid form such as, tablets, capsules, powders, solutions, suspensions, or emulsions. The compositions of the present application may be orally administered, for example, with an inert diluent, or with an assimilable edible carrier, or they may be enclosed in hard or soft shell capsules, or they may be compressed into tablets, or they may be incorporated directly with the food of the diet. For oral therapeutic administration, these compositions may be incorporated with excipients and used in the form of tablets, capsules, elixirs, suspensions, syrups, and the like. Such compositions and preparations should contain at least 0.1% of active compound. The percentage of the composition in these compositions may, of course, be varied and may conveniently be between about 2% to about 60% of the weight of the unit. Preferred compositions according to the present application are prepared so that an oral dosage unit contains between about 1 and 250 mg of active compound. The tablets, capsules, and the like may also contain a binder such as gum tragacanth, acacia, corn starch, or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, alginic acid; a lubricant such as magnesium stearate; and a sweetening agent such as sucrose, lactose, or saccharin. When the dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier, such as a fatty oil. These compositions may also be administered parenterally. Solutions or suspensions of the present compositions can be prepared in water suitably mixed with a surfactant, such as hydroxypropylcellulose. Dispersions can also be prepared in glycerol, liquid polyethylene glycols, and mixtures thereof in oils. Illustrative oils are those of petroleum, animal, vegetable, or synthetic origin, for example, peanut oil, soybean oil, or mineral oil. In general, water, saline, aqueous dextrose and related sugar solution, and glycols such as, propylene glycol or polyethylene glycol, are preferred liquid carriers, particularly for injectable solutions. Under ordinary conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms. The pharmaceutical forms suitable for injectable use include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions. In all cases, the form must be sterile and must be fluid to the extent that easy syringability exists. It must be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms, such as bacteria and fungi. The carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g., glycerol, propylene glycol, and liquid polyethylene glycol), suitable mixtures thereof, and vegetable oils. The composition of the present application may also be administered directly to the airways in the form of an aerosol. For use as aerosols, the compositions of the present application in solution or suspension may be packaged in a pressurized aerosol container together with suitable propellants, for example, hydrocarbon propellants like propane, butane, or isobutane with conventional adjuvants. The materials of the present application also may be administered in a non-pressurized form such as in a nebulizer or atomizer. The compositions of the present application further contain, in some embodiments, a second agent or pharmaceutical composition selected from the non-limiting group of anti-inflammatory agents, antidiabetic agents, hpyolipidemic agents, chemotherapeutic agents, antiviral agents, antibiotics, metabolic agents, small molecule inhibitors, protein kinase inhibitors, adjuvants, apoptotic agents, proliferative agents, organotropic targeting agents, immunological agents, antigens from pathogens, such as viruses, bacteria, fungi and parasites, optionally in the form of whole inactivated organisms, peptides, proteins, glycoproteins, carbohydrates, or combinations thereof, any examples of pharmacological or immunological agents that fall within the above-mentioned categories and that have been approved for human use that may be found in the published literature, any other bioactive component, or any combination of any of these. In some embodiments, a second agent or pharmaceutical composition selected from the non-limiting group of anti-inflammatory agents, antidiabetic agents, hpyolipidemic agents, chemotherapeutic agents, antiviral agents, antibiotics, metabolic agents, small molecule inhibitors, protein kinase inhibitors, adjuvants, apoptotic agents, proliferative agents, organotropic targeting agents. The importance of E3 ubiquitin ligases, and functional domains thereof, is highlighted by the number of normal cellular processes they regulate, and underlies the attendant diseases associated with loss of function or inappropriate targeting. See Ardley et al., “E3 Ubiquitin Ligases. A fourth aspect of the present application relates to a method of treating a disease. The method includes selecting a subject having a disease and administering the composition described herein to the subject to give the subject an increased expression level of the substrate compared to a subject not afflicted with the disease. The chimeric molecule described in this aspect is carried out in accordance with the previously described aspects of the present application. As used herein, the term “subject” refers to a mammal, such as a human, but can also be another animal such as a domestic animal (e.g., a dog, cat, or the like), a farm animal (e.g., a cow, a sheep, a pig, a horse, or the like) or a laboratory animal (e.g., a monkey, a rat, a mouse, a rabbit, a guinea pig, or the like). The term “patient” refers to a “subject” who is, or is suspected to be, afflicted with a disease or condition. The methods may involve administering compositions to a subject, where the disease possesses a measurable phenotype. The phenotype of the disease involves an increased expression level of a substrate compared to the phenotype from a subject not afflicted with the disease, in some embodiments. In this respect, chimeric molecules contained in the pharmaceutical compositions of the present application are efficacious against treating or alleviating the symptoms from a disease characterized by a phenotypic increase in the expression level of one or more substrates compared to the phenotype from a subject not afflicted with the disease. Non-limiting examples of diseases that can be treated or prevented in the context of the present application, include, cancer, metastatic cancer, solid cancers, invasive cancers, disseminated cancers, breast cancer, lung cancer, NSCLC cancer, liver cancer, prostate cancer, brain cancer, pancreatic cancer, lymphatic cancer, ovarian cancer, endometrial cancer, cervical cancer, and other solid cancers known in the art, blood cell malignancies, lymphomas, leukemias, myelomas, stroke, ischemia, myocardial infarction, congestive heart failure, stroke, ischemia, peripheral vascular disease, alcoholic liver disease, cirrhosis, Parkinson's disease, Alzheimer's disease, diabetes, cancer, arthritis, ALS, pathogenic diseases, idiopathic diseases, viral diseases, bacterial, diseases, prionic diseases, fungal diseases, parasitic diseases, arthritis, wound healing, immunodeficiency, inflammatory disease, aplastic anemia, anemia, genetic disorders, congenital disorders, type 1 diabetes, type 2 diabetes, gestational diabetes, high blood glucose, metabolic syndrome, lipodystrophy syndrome, dyslipidemia, insulin resistance, leptin resistance, atherosclerosis, vascular disease, hypercholesterolemia, hypertriglyceridemia, non-alcoholic fatty liver disease, septic shock, multiple organ dysfunction syndrome, rheumatoid arthritis, trauma, stroke, heart infarction, systemic autoimmune disease, chronic hepatitis, overweight, and/or obesity, or any combination thereof. In some embodiments, the disease is cancer, metastatic cancer, stroke, ischemia, peripheral vascular disease, alcoholic liver disease, hepatitis, cirrhosis, Parkinson's disease, Alzheimer's disease, cystic fibrosis diabetes, ALS, pathogenic diseases, idiopathic diseases, viral diseases, bacterial, diseases, prionic diseases, fungal diseases, parasitic diseases, arthritis, wound healing, immunodeficiency, inflammatory disease, aplastic anemia, anemia, genetic disorders, congenital disorders, type 1 diabetes, type 2 diabetes, gestational diabetes, high blood glucose, metabolic syndrome, lipodystrophy syndrome, dyslipidemia, insulin resistance, leptin resistance, atherosclerosis, vascular disease, hypercholesterolemia, hypertriglyceridemia, non-alcoholic fatty liver disease, overweight, or obesity, and any combination thereof. When used in vivo for therapy, the compositions are administered to the subject in effective amounts, i.e., amounts that have desired therapeutic effect. The dose and dosage regimen will depend upon the degree of the disease in the subject, the characteristics of the particular peptide used, e.g., its therapeutic index, the subject, and the subject's history. The effective amount may be determined during pre-clinical trials and clinical trials by methods familiar to physicians and clinicians. An effective amount of a peptide useful in the methods may be administered to a mammal in need thereof by any of a number of well-known methods for administering pharmaceutical compounds. Dosage, toxicity and therapeutic efficacy of the compositions can be determined by standard pharmaceutical procedures in cell cultures or experimental animals, e.g., for determining the LD50(the dose lethal to 50% of the population) and the ED50(the dose therapeutically effective in 50% of the population). The dose ratio between toxic and therapeutic effects is the therapeutic index and it can be expressed as the ratio LD50/ED50. Compounds which exhibit high therapeutic indices may be desirable. While compositions that exhibit toxic side effects may be used, care should be taken to design a delivery system that targets such compositions to the site of affected tissue in order to minimize potential damage to uninfected cells and, thereby, reduce side effects. In some embodiments, the compositions of the present application are administered orally, parenterally, subcutaneously, intravenously, intramuscularly, intraperitoneally, by intranasal instillation, by implantation, by intracavitary or intravesical instillation, intraocularly, intraarterially, intralesionally, transdermally, or by application to mucous membranes. A fifth aspect of the present application relates to a method for substrate silencing. The method includes selecting a substrate to be silenced; providing the chimeric molecule described herein; and contacting the substrate with the chimeric molecule under conditions effective to permit the formation of a substrate-molecule complex, wherein the complex mediates the degradation of the substrate to be silenced. The chimeric molecule described in this aspect is carried out in accordance with the previously described aspects of the present application. A sixth aspect of the present application relates to a method of screening agents for therapeutic efficacy against a disease. The method includes providing a biomolecule whose presence mediates a disease state; providing a test agent comprising (i) a degradation domain comprising an E3 ubiquitin ligase (E3) motif, (ii) a targeting domain capable of specifically directing the degradation domain to the biomolecule, wherein the targeting domain is heterologous to the degradation domain, and (iii) a linker coupling the degradation domain to the targeting domain; contacting the biomolecule with the test agent under conditions effective for the test agent to facilitate degradation of the biomolecule; determining the level of the biomolecule as a result of the contacting; and identifying the test agent which, based on the determining, decreases the level of the biomolecule as being a candidate for therapeutic efficacy against the disease. The chimeric molecule described in this aspect is carried out in accordance with the previously described aspects of the present application. As used herein, the terms “reference level” or “control level” refer to an amount or concentration of biomarker (or biomolecule, ligand, substrate and the like) which may be of interest for comparative purposes. In some embodiments, a reference level may be the level of at least one biomarker expressed as an average of the level of at least one biomarker taken from a control population of healthy subjects or from a diseased population possessing aberrant expression of a protein or substrate. In another embodiment, the reference level may be the level of at least one biomarker in the same subject at an earlier time, i.e., before the present assay. In even another embodiment, the reference level may be the level of at least one biomarker in the subject prior to receiving a treatment regime. As used herein, the term “sample” may include, but is not limited to, bodily tissue or a bodily fluid such as blood (or a fraction of blood such as plasma or serum), lymph, mucus, tears, saliva, sputum, urine, semen, stool, CSF, ascities fluid, or whole blood, and including biopsy samples of body tissue. A sample may also include an in vitro culture of microorganisms grown from a sample from a subject. A sample may be obtained from any subject, e.g., a subject/patient having or suspected to have a disease or condition characterized by a disease. As used herein, the term “screening” means determining whether a chimeric molecule or composition has capabilities or characteristics of preventing or slowing down (lessening) the targeted pathologic condition stated herein, namely a disease or condition characterized by defects in specified disease. As used herein, the terms “effective amount” or “therapeutically effective amount” of a chimeric molecule or composition is a quantity sufficient to achieve a desired therapeutic and/or prophylactic effect, for example, an amount which results in the prevention of or a decrease in the symptoms associated with a disease that is being treated. The amount of compound administered to the subject will depend on the type and severity of the disease and on the characteristics of the individual, such as general health, age, sex, body weight and tolerance to drugs. It will also depend on the degree, severity or stage of disease. The skilled artisan will be able to determine appropriate dosages depending on these and other factors. The term “enzyme linked immunosorbent assay” (ELISA) as used herein includes an antibody-based assay in which detection of the antigen of interest is accomplished via an enzymatic reaction producing a detectable signal. An ELISA can be run as a competitive or non-competitive format. ELISA also includes a 2-site or “sandwich” assay in which two antibodies to the antigen are used, one antibody to capture the antigen and one labeled with an enzyme or other detectable label to detect captured antibody-antigen complex. In a typical 2-site ELISA, the antigen has at least one epitope to which unlabeled antibody and an enzyme-linked antibody can bind with high affinity. An antigen can thus be affinity captured and detected using an enzyme-linked antibody. Typical enzymes of choice include alkaline phosphatase or horseradish peroxidase, both of which generate a detectable product when contacted by appropriate substrates. As used herein, the term “epitope” includes a protein determinant capable of specific binding to an antibody. Epitopes usually consist of chemically active surface groupings of molecules such as amino acids or sugar side chains and usually have specific three dimensional structural characteristics, as well as specific charge characteristics. Conformational and nonconformational epitopes are distinguished in that the binding to the former but not the latter is lost in the presence of denaturing solvents. Typically, an epitope will be a determinant region form a substrate, which can be recognized by one or more target domains. To screen for targeting domains or substrates which possess an epitope, a routine cross-blocking assay such as that described in As used herein, the term “hypervariable region” refers to the amino acid residues of an antibody which are responsible for antigen-binding. The hypervariable region generally comprises amino acid residues from a “complementarity determining region” or “CDR”, e.g., around about residues 24-34 (L1), 50-56 (L2) and 89-97 (L3) in the VL, and around about 31-35B (H1), 50-65 (H2) and 95-102 (H3) in the VH(Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991), which is hereby incorporated by reference in its entirety), and/or those residues from a “hypervariable loop” (e.g., residues 26-32 (L1), 50-52 (L2) and 91-96 (L3) in the VL, and 26-32 (H1), 52A-55 (H2) and 96-101 (H3) in the VH(Chothia and Lesk, “Canonical Structures for the Hypervariable Regions of Immunoglobulins,” As used herein, the terms “isolated” or “purified” polypeptide, peptide, molecule, or chimeric molecule, is substantially free of cellular material or other contaminating polypeptides from the cell or tissue source from which the agent is derived, or substantially free from chemical precursors or other chemicals when chemically synthesized. For example, a chimeric molecule would be free of materials that would interfere with such a molecules intended function, diagnostic or therapeutic uses. Such interfering materials may include proteins or fragments other than the materials encompassed by the chimeric molecule, enzymes, hormones and other proteinaceous and nonproteinaceous solutes. In one embodiment, the identifying is carried out with respect to a standard biomolecule level in a subject not afflicted with the disease. The identifying may also be carried out with respect to the biomolecule level absent the contacting, in some embodiments. A control level, in the regard, can be employed to compare to the level of the biomolecule in a sample. In some embodiments, the control level is the level of the biomolecule from a subject not afflicted with the disease. An overabundance of the biomolecule in the sample obtained from the subject suspected of having the disease or condition affecting substrate levels compared with the sample obtained from the healthy subject is indicative of the biomolecule-associated disease or condition in the subject being tested. There are a myriad of diseases in which the degree of overabundance of certain substrate biomolecules are known to be indicative of whether a subject is afflicted with a disease or is likely to develop a disease. See, e.g., Anderson et al., “Discovering Robust Protein Biomarkers for Disease from Relative Expression Reversals in 2-D DIGE Data,” Accordingly, the chimeric molecules and compositions of the present application are administered to a subject in need of treatment. E3 ubiquitin ligases, such as the E3 gene products encoding a E3 motif, are described above, and can be used in the present screening methods for determining the efficacy of the chimeric molecules disclosed herein. In some embodiments, suitable in vitro or in vivo assays are performed to determine the effect of the chimeric molecules and compositions of the present application and whether administration is indicated for treatment. Compositions for use in therapy can be tested in suitable animal model systems including, but not limited to rats, mice, chicken, cows, monkeys, rabbits, and the like, prior to testing in human subjects. Similarly, for in vivo testing, any of the animal model system known in the art can be used prior to administration to human subjects. Any method known to those in the art for contacting a cell, organ or tissue with a composition may be employed. In vivo methods typically include the administration of a chimeric molecule or composition, such as those described above, to a mammal, suitably a human. When used in vivo for therapy, the chimeric molecules or compositions are administered to the subject in effective amounts, as described herein. Results can be ascertained as per the empirical variables set forth at the outset of the methods described herein. In vitro methods typically include the assaying the effect of chimeric molecule or composition, such as those described above, on a sample or extract. In some embodiments, chimeric molecule efficacy can be determined by assessing the affect on substrate degradation, i.e., the ability of the chimeric molecules and compositions to exert a phenotypic change in a sample. Such methods include, but are not limited to, immunohistochemistry, immunofluorescence, ELISPOT, ELISA, or RIA. The steps of various useful immunodetection methods have been described in the scientific literature, such as, e.g., Nakamura et al., Enzyme Immunoassays: Heterogeneous and Homogeneous Systems, Handbook of Experimental Immunology, Vol. 1 Immunoassays, in their most simple and direct sense, are binding assays involving binding between antibodies and antigen. Many types and formats of immunoassays are known and all are suitable for detecting the disclosed biomarkers. Examples of immunoassays are enzyme linked immunosorbent assays (ELISAs), enzyme linked immunospot assay (ELISPOT), radioimmunoassays (RIA), radioimmune precipitation assays (RIPA), immunobead capture assays, Western blotting, dot blotting, gel-shift assays, Flow cytometry, immunohistochemistry, fluorescence microscopy, protein arrays, multiplexed bead arrays, magnetic capture, in vivo imaging, fluorescence resonance energy transfer (FRET), and fluorescence recovery/localization after photobleaching (FRAP/FLAP). In general, immunoassays involve contacting a sample suspected of containing a molecule of interest (such as the disclosed biomolecule) with an antibody to the molecule of interest or contacting an antibody to a molecule of interest (such as antibodies to the disclosed biomolecule) with a molecule that can be bound by the antibody, as the case may be, under conditions effective to allow the formation of immunocomplexes. In this regard, the skilled artisan will be able to assess the presence and or level of specific biomolecules in a given sample. Subsequently, the chimeric molecule compositions of the present application are added to the assay. Thereafter, the level of biomolecule can be assessed, i.e., the presence or level thereof, using the immunoassays described herein to determine the post-treatment phenotypic effect. Immunoassays can include methods for detecting or quantifying the amount of a biomolecule of interest in a sample, which methods generally involve the detection or quantitation of any immune complexes formed during the binding process. In general, the detection of immunocomplex formation is well known in the art and can be achieved through the application of numerous approaches. These methods are generally based upon the detection of a label or marker, such as any radioactive, fluorescent, biological or enzymatic tags or any other known label. See, for example, U.S. Pat. Nos. 3,817,837; 3,850,752; 3,939,350; 3,996,345; 4,277,437; 4,275,149 and 4,366,241, each of which is incorporated herein by reference in its entirety and specifically for teachings regarding immunodetection methods and labels. The treatment methods of the present application possess ubiquitin regions attached to targeting domains, as described above. In some embodiments, the targeting domain binds an intracellular biomolecule, as described above. Likewise, the treatment methods of the present application employ polypeptide linkers of sufficient length to prevent the steric disruption of binding between the targeting domain and the substrate. In some embodiments, the biomolecule is associated with a disease as described above. In one embodiment, the method is carried out with a plurality of test agents. A seventh aspect of the present application relates to a method of screening for disease biomarkers. The method includes providing a sample of diseased cells expressing one or more ligands; providing a plurality of chimeric molecules comprising (i) a degradation domain comprising an E3 ubiquitin ligase (E3) motif, (ii) a targeting domain capable of specifically directing the degradation domain to the one or more ligands, wherein the targeting domain is heterologous to the degradation domain, and (iii) a linker coupling the degradation domain to the targeting domain; contacting the sample with the plurality of chimeric molecules under conditions effective for the diseased cells to fail to proliferate in the absence of the chimeric molecule; determining which of the chimeric molecules permit the diseased cells to proliferate; and identifying, as biomarkers for the disease, based on the determining the ligands which bind to the chimeric molecules and permit diseased cells to proliferate. The chimeric molecule described in this aspect is carried out in accordance with the previously described aspects of the present application. As used herein, the terms “biomarker” or “biomolecule” or “molecule” refer to a polypeptide (of a particular expression level) which is differentially present in a sample taken from patients having a disease as compared to a comparable sample taken from a control subject or a population of control subjects. As used herein, the terms “ligand” or “substrate” refer to substance that are able to bind to and form transient or stable complexes with a protein, molecule, chimeric molecule, ligand (dimer), substrate (dimer), a second substrate, a second ligand, target domain, regions, potions, and fragments thereof, ubiquitin or E3 motif regions, domains, or portions thereof, biomolecules, biomarkers, and the like, to serve a biological purpose, for example a substrate which interacts with an enzyme in the process of an enzymatic reaction. Ligands also include signal triggering molecules which bind to sites on a target protein, by intermolecular forces such as ionic bonds, hydrogen bonds and Van der Waals forces. In some embodiments, substrates bind ligands and/or ligands bind substrates. Many, if not all diseases, are complex and multifactorial. When considering neurodegeneration, for example, substantial neuronal cell loss occurs before pathologic presentation. Screening for and developing such drugs—to treat neurodegenerative diseases—is further stymied by ancillary therapies which ameliorate the symptoms. Thus, target detection is obfuscated by prior therapeutic administration, which, may in turn, slow disease progression and further confound treatment regimes. In this way, the present application provides new, inventive, screening methods for elucidation of disease biomarkers by employing phenotypic screening analyses. See, e.g., Pruss, R. M., “Phenotypic Screening Strategies for Neurodegenerative Diseases: A Pathway to Discover Novel Drug Candidates and Potential Disease Targets or Mechanisms.” Phenotypic screening involves using an appropriate sample, e.g., class of cells, cell extract, neurons, tissue, and the like, from a patient afflicted with a disease and subjecting the sample to one or more chimeric molecules as described herein. Subsequently, the sample is screened for viability, proliferation, cell processes and/or phenotypic characteristic of the diseased cell, e.g., shrinking, loss of membrane potential, morphological changes, and the like. Image analysis software allows for cell bodies or other objects to empirically assess the results. Hits coming from the screen may maintain cell survival by stimulating survival pathways, mimicking trophic factors, or inhibiting death signaling. Higher content screening and profiling in target-directed secondary assays can then be used to identify targets and mechanisms of action of promising hits. Examples of diseases conditions from which a biomarker screening analysis can be performed include the diseases described above. In some embodiments, the method of screening for disease biomarkers includes a plurality of molecules, where the molecules possess a E3 motif as described above. In some embodiments, the biomarker screening methods include molecules possessing a targeting domain as described above. The screening methods of the present application employ polypeptide linkers of sufficient length to prevent the steric disruption of binding between the targeting domain and the biomolecule and/or ligand. Once a chimeric molecule is determined to provide a therapeutic indication, the biomarker is isolated using the targeting domain region (or the entire chimeric molecule) to immunoprecipitate the biomarker, from a sample, which is subsequently identified using methods well known in the art. Biomarker isolation and purification methods include, but are not limited to, for example, HPLC or FPLC chromatography using size-exclusion or affinity-based column resins. See, e.g., Sambrook et al. 1989, Cold Spring Harbor Laboratory Press, which is hereby incorporated by reference in its entirety. Active fragments, derivatives, or variants of the polypeptides of the present application may be recognized by, for example, the deletion or addition of amino acids that have minimal influence on the properties, secondary structure, and biological activity of the polypeptide. For example, a polypeptide may be joined to a signal (or leader) sequence at the N-terminal end of the protein which co-translationally or post-translationally directs sub-cellular or extracellular localization of the protein. The biomarker can then be elucidated using techniques known in the art. In some embodiments, determining the identity of the biomarker is performed using MALDI-TOF, mass spectrometry, mass spectroscopy, protein sequencing, antibody interactions, western blot, immunoassay, ELISA, chromatographic techniques, reverse proteomics, immunoprecipitations, radioimmunoassay, and immunofluorescence, or any combinations thereof. Suitable mass spectrometric techniques for the study and identification of proteins include, laser desorption ionization mass spectrometry and electrospray ionization mass spectrometry. Within the category of laser desorption ionization (LDI) mass spectrometry (MS), both matrix assisted LDI (MALDI) and surface assisted LDI (SELDI) time-of-flight (TOF) MS may be employed. SELDI TOF-MS is particularly well-suited for use in the present methods because it provides attomole sensitivity for analysis, quantification of low abundant proteins (pg-ng/ml) and highly reproducible results. The methods described herein can be performed, e.g., by utilizing pre-packaged kits comprising at least one reagent, e.g., a chimeric molecule or composition described herein, which can be conveniently used, e.g., in clinical settings to treat subjects exhibiting symptoms of a disease or illness involving an overexpressed substrate, biomolecule, or biomarker. Furthermore, any cell type or tissue in which the chimeric molecule of the present application can be expressed is suitable for use in the kits described herein. In another aspect of the present application, a kit or reagent system for using the chimeric molecules and compositions of the present application. Such kits will contain a reagent combination including the particular elements required to conduct an assay according to the methods disclosed herein. The reagent system is presented in a commercially packaged form, as a composition or admixture where the compatibility of the reagents will allow, in a test device configuration, or more typically as a test kit, i.e., a packaged combination of one or more containers, devices, or the like holding the necessary reagents, and preferably including written instructions for the performance of assays. The kit may be adapted for any configuration of an assay and may include compositions for performing any of the various assay formats described herein. Reagents useful for the disclosed methods can be stored in solution or can be lyophilized. When lyophilized, some or all of the reagents can be readily stored in microtiter plate wells for easy use after reconstitution. It is contemplated that any method for lyophilizing reagents known in the art would be suitable for preparing dried down reagents useful for the disclosed methods. Also within the scope of the present application are kits comprising the chimeric molecules/compositions and second agents of the application and instructions for use. The kits are useful for detecting the presence of a substrate in a biological sample e.g., any body fluid including, but not limited to, e.g., serum, plasma, lymph, cystic fluid, urine, stool, cerebrospinal fluid, acitic fluid or blood and including biopsy samples of body tissue. For example, the kit can comprise one or more chimeric molecules composed of a E3 motif ubiquitin region linked to a targeting domain capable of binding a substrate in a biological sample. The following examples are provided to illustrate embodiments of the present application but they are by no means intended to limit its scope. The following examples are included to demonstrate illustrative embodiments of the present application. It should be appreciated by those of skill in the art that the techniques disclosed in the examples which follow represent techniques discovered by the inventors to function well in the practice of the present application, and thus can be considered to constitute preferred modes for its practice. However, those of skill in the art should, in light of the present application, appreciate that many changes can be made in the specific embodiments which are disclosed and still obtain a like or similar result without departing from the spirit and scope of the present application. Plasmids. All plasmids used in this study are provided in Table 1. For creation of GFP-directed uAbs, plasmid pcDNA3-HF-GS2 was created by PCR amplification of GS2 from pHFT2-GS2 (Koide et al., “Teaching an Old Scaffold New Tricks: Monobodies Constructed Using Alternative Surfaces of the FN3 Scaffold,” Plasmid pET24d-GS2-IpaH9.8 and pET24d-IpaH9.8ΔLRR were created by PCR amplification of full-length GS2-IpaH9.8 and truncated IpaH9.8ΔLRR, respectively, with primers that introduced NcoI and NotI (GS2-IpaH9.8) or NheI and NotI (IpaH9.8 ΔLRR) sites, after which the resulting PCR products were ligated into pET24d(+). Plasmid pET28a-GS2 was created by PCR amplification of GS2 from pHFT2-GS2 using primers that introduced an upstream NcoI site and downstream FLAG, 6×-His, and HindIII sequences, after which the resulting PCR product was ligated into pET28a(+). Plasmid pTriEx-3-GS2-IpaH9.8C337Awas created by PCR amplification of GS2-IpaH9.8C337Afrom pcDNA3-GS2-IpaH9.8C337Awith primers that introduced EcoRV and HindIII sites, after which the resulting PCR product was ligated into pTriEx-3. Plasmid pET28a-EGFP was created by PCR amplification of GFP with primers adding C-terminal 6×-His tag, after which the resulting PCR product was ligated in pET28a(+). All plasmids were verified by DNA sequencing at the Cornell Biotechnology Resource Center (BRC). Cell Lines, Culture and Transfection. All cell lines used in this study are provided in Table 1. Briefly, HEK293T and HeLa cells were obtained from ATCC, HeLa H2B-EGFP cells were kindly provided by Elena Nigg, and MCF10A rtTA cells were kindly provided by Matthew Paszek, HEK293T, HeLa, and HeLa H2B-EGFP cells were cultured in DMEM with 4.5 g/L glucose and L-glutamine (VWR) supplemented with 10% FetalCloneI (VWR) and 1% penicillin-streptomycin-amphotericin B (ThermoFisher) MCF-10a cells were grown in DMEM/F12 media (ThermoFisher) supplemented with 5% horse serum (ThermoFisher), 20 ng/mL EGF (Peprotech), 0.5 mg/mL hydrocortisone (Sigma), 100 ng/mL cholera toxin (Sigma), 10 μg/mL insulin (Sigma), and 1% penicillin-streptomycin-amphotericin B (ThermoFisher). All cells were maintained at 37° C., 5% CO2and and 90% relative humidity (RH). Stable MCF10A rtTA cell lines were generated using Nucleofection Kit V (Lonza) and HyPBase, an expression plasmid for the hyperactive version of the PiggyBac transposase. Transposition of MCF10A rtTA cells was performed to generate the following stable lines: MCF10A EGFP-HRasG12V; MCF10A EGFP-HRasG12V:GS2-IpaH9.8; MCF10A EGFP-HRasG12V:GS2-IpaH9.8C337A; and MCF10A GS2-IpaH9.8. Stable cell lines were selected using 200 μg/mL hygromycin B (ThermoFisher). Stable HEK293T cell lines expressing EGFP, EGFP-HRasG12V, ERK2-EGFP, d2EGFP were generated by lentiviral transformation. Specifically, pLV IRES eGFP, pcDH1 eGFP-HRasG12V, pcDH1 ERK2-EGFP, or pHIV-d2EGFP were transfected into HEK293T cells along with psPAX2 and pMD2.G by calcium phosphate transfection. Media was replaced after ˜16 h, followed by a 48-h incubation to allow virus production. Viral supernatant was removed and Polybrene (Sigma-Aldrich) added to a final concentration of 8 μg/mL, followed by clearance of cell debris by centrifugation at 2,000 rpm for 5 min. Resultant supernatant was diluted 1:6 with cell media and added to previously plated HEK293T cells for stable integration. HEK293T EGFP and HEK293T ERK2-EGFP cell lines were selected by fluorescence activated cell sorting (BD FACSAria). The HEK293T EGFP-HRasG12Vcell line was selected using 1 μg/mL puromycin (Sigma-Aldrich). Western Blot Analysis. HEK293T cells were plated at 10,000 cells/cm2 and transfected as described above before lysis with RIPA lysis buffer (Thermo Fisher). MCF10A cells were plated at 20,000 cells/cm2 and induced with 0.2 μg/mL doxycycline for 24 h before lysis with cell lysis buffer. Lysates were separated on Any kD polyacrylamide gels (Bio-Rad) and transferred to PVDF membranes. α-HIS-HRP (Abcam), α-GFP (Krackeler) and α-GAPDH (Millipore) antibodies were diluted 1:5,000 and in TBST+1% milk and incubated for 1 h at room temperature. Secondary antibody goat anti-mouse IgG with HRP conjugation (Promega) was diluted at 1:2,500 and used as needed. Flow Cytometric Analysis. Cells were passed into 12-well plates at 10,000 cells/cm2. 16-24 h after seeding, cells were transiently transfected with 1 μg total DNA at a 1:2 ratio of DNA:jetPrime (Polyplus Transfection). Cells were transfected with 0.05 μg of target, 0.25 μg of ubiquibody or control, and balanced with empty pcDNA3 vector. Culture media was replaced 4-6 h post-transfection. Then, 24 h post-transfection, cells were harvested and resuspended in phosphate buffered saline (PBS) for analysis using a FACSCalibur or FACSAria Fusion (BD Biosciences). FlowJo Version 10 was used to analyze samples by geometric mean fluorescence determined from 10,000 events. Microscopy. Cells were plated at 10,000 cells/cm2on a glass bottom 12-well plate pre-treated with poly-L-lysine (Sigma-Aldrich). After seeding for 16-24 h, cells were transfected with 1 μg total DNA at a 1:2 ratio of DNA:jetPrime (Polyplus Transfection). Cells were transfected with 0.05 μg of target, 0.25 μg of ubiquibody or control, and balanced with empty pcDNA3 vector. Culture media was replaced 4-6 h post-transfection. Then, 24 h post-transfection, cells were fixed with 4% paraformaldehyde. For EGFR-EGFP samples, cells were blocked with 5% normal goat serum in PBS for 2 h at room temperature. The anti-EGFR antibody (Cell Signalling #4267) was diluted 1:200 in 5% normal goat serum in PBS and incubated overnight at 4° C. Cells were washed three times with PBS, then incubated for 1 h at room temperature with anti-rabbit-AF647 diluted 1:200 in 5% normal goat serum in PBS. Cells were washed three times with PBS. Cell nuclei were stained with Hoescht diluted 1:10,000 in PBS for 10 min, then washed three times in PBS. Samples were imaged on an inverted Zeiss LSM88-confocal/multiphoton microscope (i880) using a 40× water immersion objective. Images were analyzed with FIJI. Protein Expression and Purification. Purified proteins were obtained by growing ELISA. A 96-well enzyme immunoassay plate was coated with 100 μL of EGFP at 10 μg/mL in 0.05 M NaCO3buffer, pH 9.6 at 4° C. overnight. The plate was then washed three times 200 μL PBST (1×PBS+0.1% Tween 20) per well and blocked with 250 μL PBS with 3% milk per well at room temperature for 3 h, slowly mixing. The plate was washed three more times, followed by the addition of serial dilutions of purified proteins in blocking buffer at 60 μL per well. Plate was incubated at room temperature, slowly mixing for 1 h. The plate was washed three times to remove un-bound protein and then incubated with 50 μL/well of anti-FLAG (DDDYK) antibody conjugated to horseradish peroxidase (HRP) diluted 1:10,000 in PBST+1% milk for 1 h with slow mixing. The plate was washed three times before the addition of 50 μL/well 1-Step Ultra TMB (3,3′,5,5′-tetramethylbenzidine) (ThermoFisher). The reaction was allowed to incubate with slow mixing and then quenched with 50 μL/well of 3N H2SO4. The quenched plate was then read at 450 nm. Synthesis and Characterization of Cationic Polypeptides. N4 (TEP) polyamines were synthesized as the researchers of the present group described recently (Li et al., “Polyamine-Mediated Stoichiometric Assembly of Ribonucleoproteins for Enhanced mRNA Delivery,” Preparation of mRNA by In Vitro Transcription. cDNA encoding uAbs was cloned into pGEM4Z/GFP/A64 by replacing the GFP fragment with XbaI and NotI sites. Additionally, the human α-globin 3′ UTR sequence was placed between the cDNA and the poly A tail using NotI and EcoRI to improve mRNA translation. Linearization with SpeI, followed by in vitro transcription (IVT) with HiScribe™ T7 High Yield RNA Synthesis Kit (NEB), yielded a transcript containing 64 nucleotides of vector-derived sequence, the coding sequence, α-globin 3′ UTR, and 64 A residues. In a typical 20 μl reaction, the following nucleotides were prepared: ATP (10 mM), pseudo-UTP (10 mM), methyl-CTP (10 mM), GTP (2 mM), anti-reverse Cap analog (8 mM, NEB). RNA was purified by RNeasy purification kit (Qiagen, Hilden, Germany). RNA quality was confirmed by running a 1% agarose gel. Concentration was determined by Abs260. Nanoplex Transfection. Polyamines were dissolved in 10 mM HEPES buffer (pH 7.4). For each well of a 96-well plate, 200 ng mRNA diluted in 5 μl OptiMEM (Thermo Fisher) was mixed with 5 μl OptiMEM containing PABP (mRNA:PABP weight ratio=1:5) at room temperature for 10 min. Afterwards, 5 μl OptiMEM containing polyamines was added and incubated at room temperature for 15 min prior to transfection into HEK293T stably expressing d2EGFP. Polyamines were adjusted to achieve 50 to 1 (N/P) ratio for transfection. EGFP expression was measured by BD FACSCelesta (Becton Dickinson) at different time points after transfection. Animal Experiments. Mouse care and experimental procedures were performed under pathogen-free conditions in accordance with established institutional guidelines and approved protocols from the MIT Division of Comparative Medicine. C57BL/6-Tg(UBC-GFP)30Scha/J mice were purchased from Jackson Laboratory. 8-10 week-old mice were injected subcutaneously in ears with 5 μg mRNA and 25 μg PABP packaged with N4 (TEP) polyamines in a volume of 25 μl OptiMEM under anesthesia. Fluorescent imaging was performed with a CCD camera mounted in a light-tight specimen box (Xenogen). The exposure time was 1 s. Imaging and quantification of signals were controlled by Living Image acquisition and analysis software (Xenogen). To determine whether E3 ubiquitin ligase mimics from pathogenic bacteria could be redesigned for silencing of non-native targets, the focus was on a panel of 14 candidate enzymes representing the major classes of E3s found in bacteria to date (Table 2, infra). Maculins et al., “Bacteria-Host Relationship: Ubiquitin Ligases as Weapons of Invasion. Abbreviations in Table 2: NEL, novel E3 ligase; HECT, homologous to E6AP C terminus; SCF, Skp1/Cdc53 or Cullen-1/F-box protein; SPOP, speckle-type POZ protein; VHL, von Hippel-Lindau; ECV, Elongin B/C, Cullen-2, VHL
This panel included E3 mimics with folds similar to eukaryotic E3s such as HECT-type, RING or U-box (RING/U-box)-type, and F-box domains, as well as unconventional E3s with folds unlike any other eukaryotic E3s such as novel E3 ligase (NEL), XL-box-containing, and SidC. In general, uAbs were engineered by removing the native substrate-binding domain from each E3 mimic and replacing it with a synthetic binding protein ( In light of this robust silencing activity, the attention was focused on the GS2-IpaH9.8. In cells expressing this chimera, the elimination of EGFP was efficient, with removal of up to 90% of the fluorescence activity ( To benchmark the potency of the present engineered bacterial ligase, the GFP silencing activity catalyzed by GS2-IpaH9.8 was compared with that of other synthetic ligases based on eukaryotic E3 machinery that have previously been reconfigured for targeted proteolysis. Zhou et al., “Harnessing the Ubiquitination Machinery to Target the Degradation of Specific Cellular Proteins,” To more deeply explore the substrate compatibility issue, the ability of GS2-IpaH9.8 to degrade a range of different substrates was tested. A growing number of GFP-derived fluorescent proteins (FPs) have been developed and optimized over the years, providing a diverse collection of new tools for biological imaging. Tsien, R. Y., “The Green Fluorescent Protein,” Encouraged by the ability of GS2-IpaH9.8 to degrade different FPs, the ability of GS2-IpaH9.8 to degrade structurally diverse, FP-tagged substrate proteins was next evaluated. GS2-IpaH9.8 proficiently degraded 15 unique target proteins that varied in terms of their molecular weight (27-179 kDa) and subcellular localization (i.e., cytoplasm, nucleus, membrane-associated, and transmembrane) ( An attractive feature of uAbs is their highly modular architecture—the E3 catalytic domain and synthetic binding protein domain can be interchanged to reprogram the activity and specificity. Indeed, the results above revealed the ease with which different bacterial and eukaryotic E3 domains can be chimerized to form functional uAbs. To investigate the interchangeability of the synthetic binding protein domain in IpaH9.8-based uAbs, GS2 was first replaced with other high-affinity GFP-binding proteins such as the FN3 monobody GS5 (Kd=62 nM) (Koide et al., “Teaching an Old Scaffold New Tricks: Monobodies Constructed Using Alternative Surfaces of the FN3 Scaffold,” Next, the compatibility of the IpaH9.8 catalytic domain with two different FN3 monobodies was investigated: NSa5 that is specific for the Src-homology 2 (SH2) domain of SHP2 (Sha et al., “Dissection of the BCR-ABL Signaling Network Using Highly Specific Monobody Inhibitors to the SHP2 SH2 Domains,” In all the experiments described above, efficient knockdown was achieved when GS2-IpaH9.8 and its corresponding target were transiently expressed. However, transient expression is not always an option, due to the experimental timescale, necessity for a precise expression profile, or the use of a recalcitrant mammalian cell line. Thus, to demonstrate the flexibility of GS2-IpaH9.8-mediated silencing, degradation activity was evaluated against target proteins that were expressed as stably integrated transgenes. Specifically, when GS2-IpaH9.8 was transiently expressed in cells that stably co-expressed EGFP, reduction of fluorescence activity was virtually identical to that observed for transiently expressed EGFP ( From a therapeutic standpoint, one of the biggest challenges facing protein-based technologies such as uAbs is intracellular delivery. Osherovich, L., “Degradation From Within,” Encouraged by these results, uAb nanoplex-mediated delivery and silencing activity in vivo was next evaluated. Transgenic UBI-GFP/BL6 mice, which constitutively express EGFP in all tissues (Schaefer et al., “Observation of Antigen-Dependent CD8+ T-Cell/Dendritic Cell Interactions Ubiquibodies are a relatively new proteome editing modality that enable selective removal of otherwise stable proteins in somatic cells (Portnoff et al., “Ubiquibodies, Synthetic E3 Ubiquitin Ligases Endowed With Unnatural Substrate Specificity for Targeted Protein Silencing,” While the work here was predominantly focused on silencing FPs and FP-tagged substrates, IpaH9.8-based uAbs that potently degraded disease-related targets including HRas was designed, which together with KRas and NRas comprise the most commonly mutated oncoproteins in cancer, and SHP2, a regulator of the Ras/MAPK signaling pathway. Importantly, the ability to deplete these clinically important targets along with all of the other FP fusions serves to highlight the extraordinary modularity of the uAb technology. Simply swapping the native substrate-binding domain of the E3 ubiquitin ligase can generate a made-to-order uAb with specificity for a different substrate protein. Interestingly, From a drug development standpoint, pharmacological control of gene products has traditionally been achieved using small molecule inhibitors that target enzymes and receptors having well-defined hydrophobic pockets where the small molecules are tightly bound. Unfortunately, a majority (˜80-85%) of the human proteome is comprised of intractable targets, such as transcription factors, scaffold proteins, and non-enzymatic proteins, that cannot be inhibited pharmacologically and thus have been deemed ‘undruggable’. Crews, C. M., “Targeting the Undruggable Proteome: The Small Molecules of My Dreams,” A major advantage of uAbs is the ease with which they can be rapidly adapted to hit a variety of intracellular targets due to their recombinant, modular design, which capitalizes on a large, preexisting repertoire of synthetic binding proteins as well as systematic, genome-wide efforts to generate and validate protein binders de novo against the human proteome. Colwill et at., “A Roadmap to Generate Renewable Protein Binders to the Human Proteome,” Although preferred embodiments have been depicted and described in detail herein, it will be apparent to those skilled in the relevant art that various modifications, additions, substitutions, and the like can be made without departing from the spirit of the application and these are therefore considered to be within the scope of the present application as defined in the claims which follow. The present application relates to an isolated chimeric molecule comprising a degradation domain comprising an E3 ubiquitin ligase (E3) motif and a targeting domain capable of specifically directing the degradation domain to a substrate, where the targeting domain is heterologous to the degradation domain. A linker couples the degradation domain to the targeting domain. Also disclosed are compositions as well as methods of treating a disease, substrate silencing, screening agents for therapeutic efficacy against a disease, and methods of screening for disease biomarkers. 1. An isolated chimeric molecule comprising:
a degradation domain comprising an E3 ubiquitin ligase (E3) motif; a targeting domain capable of specifically directing said degradation domain to a substrate, wherein said targeting domain is heterologous to said degradation domain; and a linker coupling said degradation domain to said targeting domain. 2. The chimeric molecule of 3. The chimeric molecule of 4. The chimeric molecule of 5. The chimeric molecule of 6. The chimeric molecule of 7. The chimeric molecule of 8. The chimeric molecule of 9. The chimeric molecule of 10. The chimeric molecule of 11. The chimeric molecule of 12. The chimeric molecule of 13. The chimeric molecule of 14. The chimeric molecule of 15. The chimeric molecule of 16. The chimeric molecule of 17. The chimeric molecule of 18. A method of forming a ribonucleoprotein comprising:
providing a mRNA encoding the isolated chimeric molecule of providing one or more polyadenosine binding proteins (“PABP”); and assembling a ribonucleoprotein complex from the mRNA and the one or more PABPs. 19. The method of 20. A composition comprising:
the chimeric molecule of a pharmaceutically-acceptable carrier. 21. The composition of a second agent selected from the group consisting of an anti-inflammatory agent, an antidiabetic agent, a hypolipidemic agent, a chemotherapeutic agent, an antiviral agent, an antibiotic, a metabolic agent, a small molecule inhibitor, a protein kinase inhibitor, adjuvants, apoptotic agents, a proliferative agent, and organotropic targeting agents, and any combination thereof. 22. A method of treating a disease comprising:
selecting a subject having a disease and administering the composition of 23. The method of 24. The method of 25. A method for substrate silencing, the method comprising:
selecting a substrate to be silenced; providing said chimeric molecule of contacting said substrate with said chimeric molecule under conditions effective to permit the formation of a substrate-molecule complex, wherein said complex mediates the degradation of said substrate to be silenced. 26. The method of 27. The method of 28. A method of screening agents for therapeutic efficacy against a disease, said method comprising:
providing a biomolecule whose presence mediates a disease state; providing a test agent comprising (i) a degradation domain comprising an E3 ubiquitin ligase (E3) motif, (ii) a targeting domain capable of specifically directing said degradation domain to said biomolecule, wherein said targeting domain is heterologous to said degradation domain, and (iii) a linker coupling said degradation domain to said targeting domain; contacting said biomolecule with said test agent under conditions effective for the test agent to facilitate degradation of the biomolecule; determining the level of said biomolecule as a result of said contacting; and identifying said test agent which, based on said determining, decreases the level of said biomolecule as being a candidate for therapeutic efficacy against said disease. 29. The method of 30. The method of 31. The method of 32. The method of 33. The method of 34. The method of 35. The method of 36. The method of 37. The method of 38. The method of 39. The method of 40. The method of 41. The method of 42. The method of 43. The method of 44. A method of screening for disease biomarkers, said method comprising:
providing a sample of diseased cells expressing one or more ligands; providing a plurality of chimeric molecules comprising (i) a degradation domain comprising an E3 ubiquitin ligase (E3) motif, (ii) a targeting domain capable of specifically directing said degradation domain to said one or more ligands, wherein said targeting domain is heterologous to said degradation domain, and (iii) a linker coupling said degradation domain to said targeting domain; contacting said sample with said plurality of chimeric molecules under conditions effective for the diseased cells to fail to proliferate in the absence of the chimeric molecule; determining which of said chimeric molecules permit the diseased cells to proliferate; and identifying, as biomarkers for the disease, based on said determining the ligands which bind to the chimeric molecules and permit diseased cells to proliferate. 45. The method of 46. The method of 47. The method of 48. The method of 49. The method of 50. The method of 51. The method of 52. The method of 53. The method of FIELD
BACKGROUND
SUMMARY
BRIEF DESCRIPTION OF THE DRAWINGS
DETAILED DESCRIPTION
MAGINRAGPSGAYFVGHTDPEPVSGQAHGSGSGASSSNSPQVQPRPSNTPP SNAPAPPPTGRERLSRSTALSRQTREWLEQGMPTAEDASVRRRPQVTADAA TPRAEARRTPEATADASAPRRGAVAHANSIVQQLVSEGADISHTRNMLRNA MNGDAVAFSRVEQNIFRQHFPNMPMHGISRDSELAIELRGALRRAVHQQAA SAPVRSPTPTPASPAASSSGSSQRSLFGRFARLMAPNQGRSSNTAASQTPV DRSPPRVNQRPIRVDRAAMRNRGNDEADAALRGLVQQGVNLEHLRTALERH VMQRLPIPLDIGSALQNVGINPSIDLGESLVQHPLLNLNVALNRMLGLRPS AERAPRPAVPVAPATASRRPDGTRATRLRVMPEREDYENNVAYGVRLLNLN PGVGVRQAVAAFVTDRAERPAVVANIRAALDPIASQFSQLRTISKADAESE ELGFKDAADHHTDDVTHCLFGGELSLSNPDQQVIGLAGNPTDTSQPYSQEG NKDLAFMDMKKLAQFLAGKPEHPMTRETLNAENIAKYAFRIVP ATGGCGGGTATCAATAGAGCGGGACCATCGGGCGCTTATTTTGTTGGCCAC ACAGACCCCGAGCCAGTATCGGGGCAAGCACACGGATCCGGCAGCGGCGCC AGCTCCTCGAACAGTCCGCAGGTTCAGCCGCGACCCTCGAATACTCCCCCG TCGAACGCGCCCGCACCGCCGCCAACCGGACGTGAGAGGCTTTCACGATCC ACGGCGCTGTCGCGCCAAACCAGGGAGTGGCTGGAGCAGGGTATGCCTACA GCGGAGGATGCCAGCGTGCGTCGTAGGCCACAGGTGACTGCCGATGCCGCA ACGCCGCGTGCAGAGGCAAGACGCACGCCGGAGGCAACTGCCGATGCCAGC GCACCGCGTAGAGGGGCGGTTGCACACGCCAACAGTATCGTTCAGCAATTG GTCAGTGAGGGCGCTGATATTTCGCATACTCGTAACATGCTCCGCAATGCA ATGAATGGCGACGCAGTCGCTTTTTCTCGAGTAGAACAGAACATATTTCGC CAGCATTTCCCGAACATGCCCATGCATGGAATCAGCCGAGATTCGGAACTC GCTATCGAGCTCCGTGGGGCGCTTCGTCGAGCGGTTCACCAACAGGCGGCG TCAGCGCCAGTGAGGTCGCCCACGCCAACACCGGCCAGCCCTGCGGCATCA TCATCGGGCAGCAGTCAGCGTTCTTTATTTGGACGGTTTGCCCGTTTGATG GCGCCAAACCAGGGACGGTCGTCGAACACTGCCGCCTCTCAGACGCCGGTC GACAGGAGCCCGCCACGCGTCAACCAAAGACCCATACGCGTCGACAGGGCT GCGATGCGTAATCGTGGCAATGACGAGGCGGACGCCGCGCTGCGGGGGTTA GTACAACAGGGGGTCAATTTAGAGCACCTGCGCACGGCCCTTGAAAGACAT GTAATGCAGCGCCTCCCTATCCCCCTCGATATAGGCAGCGCGTTGCAGAAT GTGGGAATTAACCCAAGTATCGACTTGGGGGAAAGCCTTGTGCAACATCCC CTGCTGAATTTGAATGTAGCGTTGAATCGCATGCTGGGGCTGCGTCCCAGC GCTGAAAGAGCGCCTCGTCCAGCCGTCCCCGTGGCTCCCGCGACCGCCTCC AGGCGACCGGATGGTACGCGTGCAACACGATTGCGGGTGATGCCGGAGCGG GAGGATTACGAAAATAATGTGGCTTATGGAGTGCGCTTGCTTAACCTGAAC CCGGGGGTGGGGGTAAGGCAGGCTGTTGCGGCCTTTGTAACCGACCGGGCT GAGCGGCCAGCAGTGGTGGCTAATATCCGGGCAGCCCTGGACCCTATCGCG TCACAATTCAGTCAGCTGCGCACAATTTCGAAGGCCGATGCTGAATCTGAA GAGCTGGGTTTTAAGGATGCGGCAGATCATCACACGGATGACGTGACGCAC TGTCTTTTTGGCGGAGAATTGTCGCTGAGTAATCCGGATCAGCAGGTGATC GGTTTGGCGGGTAATCCGACGGACACGTCGCAGCCTTACAGCCAAGAGGGA AATAAGGACCTGGCGTTCATGGATATGAAAAAACTTGCCCAATTCCTCGCA GGCAAGCCTGAGCATCCGATGACCAGAGAAACGCTTAACGCCGAAAATATC GCCAAGTATGCTTTTAGAATAGTCCCC MKPAHNPSFFRSFCGLGCISRLSVEEQNITDYHRIWDNWAKEGAATEDRTQ AVRLLKICLAFQEPALNLSLLRLRSLPYLPPHIQELNISSNELRSLPELPP SLTVLKASDNRLSRLPALPPHLVALDVSLNRVLTCLPSLPSSLQSLSALLN SLETLPDLPPALQKLSVGNNQLTALPELPCELQELSAFDNRLQELPPLPQN LRLLNVGENQLHRLPELPQRLQSLYIPNNQLNTLPDSIMNLHIYADVNIYN NPLSTRTLQALQRLTSSPDYHGPRIYFSMSDGQQNTLHRPLADAVTAWFPE NKQSDVSQIWHAFEHEEHANTFSAFLDRLSDTVSARNTSGFREQVAAWLEK LSASAELRQQSFAVAADATESCEDRVALTWNNLRKTLLVHQASEGLFDNDT GALLSLGREMFRLEILEDIARDKVRTLHFVDEIEVYLAFQTMLAEKLQLST AVKEMRFYGVSGVTANDLRTAEAMVRSREENEFTDWFSLWGPWHAVLKRTE ADRWALAEEQKYEMLENEYPQRVADRLKASGLSGDADAEREAGAQVMRETE QQIYRQLTDEVLALRLPENGSQUIHS ATGAAACCTGCCCACAATCCTTCTTTTTTCCGCTCCTTTTGTGGTTTAGGA TGTATATCCCGTTTATCCGTAGAAGAGCAAAATATCACGGATTATCACCGC ATCTGGGATAACTGGGCCAAGGAAGGTGCTGCAACAGAAGACCGAACACAG GCAGTTCGATTACTGAAAATATGTCTGGCTTTTCAAGAGCCAGCCCTCAAT TTAAGTTTACTCAGATTACGCTCTCTCCCATACCTGCCCCCGCACATACAA GAACTTAACATCTCTAGCAATGAGCTACGCTCTCTGCCAGAACTCCCTCCG TCCTTAACTGTACTTAAAGCCAGCGATAACAGACTGAGCAGGCTCCCGGCT CTTCCGCCTCACCTGGTCGCTCTTGATGTTTCACTTAACAGAGTTTTAACA TGTTTGCCTTCTCTTCCATCTTCCTTGCAGTCACTCTCAGCCCTTCTCAAT AGCCTGGAGACGCTACCTGATCTTCCCCCGGCTCTACAAAAACTTTCTGTT GGCAACAACCAGCTTACTGCCTTACCAGAATTACCATGTGAACTACAGGAA CTAAGTGCTTTTGATAACAGATTACAAGAGCTACCGCCCCTTCCTCAAAAT CTGAGGCTTTTAAACGTTGGGGAAAACCAACTACACAGACTGCCCGAACTT CCACAACGTCTGCAATCACTATATATCCCTAACAATCAGCTGAACACATTG CCAGACAGTATCATGAATCTGCACATTTATGCAGATGTTAATATTTATAAC AATCCATTGTCGACTCGCACTCTGCAAGCCCTGCAAAGATTAACCTCTTCG CCGGACTACCACGGCCCACGGATTTACTTCTCCATGAGTGACGGACAACAG AATACACTCCATCGCCCCCTGGCTGATGCCGTGACAGCATGGTTCCCGGAA AACAAACAATCTGATGTATCACAGATATGGCATGCTTTTGAACATGAAGAG CATGCCAACACCTTTTCCGCGTTCCTTGACCGCCTTTCCGATACCGTCTCT GCACGCAATACCTCCGGATTCCGTGAACAGGTCGCTGCATGGCTGGAAAAA CTCAGTGCCTCTGCGGAGCTTCGACAGCAGTCTTTCGCTGTTGCTGCTGAT GCCACTGAGAGCTGTGAGGACCGTGTCGCGCTCACATGGAACAATCTCCGG AAAACCCTCCTGGTCCATCAGGCATCAGAAGGCCTTTTCGATAATGATACC GGCGCTCTGCTCTCCCTGGGCAGGGAAATGTTCCGCCTCGAAATTCTGGAG GACATTGCCCGGGATAAAGTCAGAACTCTCCATTTTGTGGATGAGATAGAA GTCTACCTGGCCTTCCAGACCATGCTCGCAGAGAAACTTCAGCTCTCTACT GCCGTGAAGGAAATGCGTTTCTATGGCGTGTCGGGAGTGACAGCAAATGAC CTCCGCACTGCCGAAGCCATGGTCAGAAGCCGTGAAGAGAATGAATTTACG GACTGGTTCTCCCTCTGGGGACCATGGCATGCTGTACTGAAGCGTACGGAA GCTGACCGCTGGGCGCTGGCAGAAGAGCAGAAATATGAGATGCTGGAGAAT GAGTACCCTCAGAGGGTGGCTGACCGGCTGAAAGCATCAGGTCTGAGCGGT GATGCGGATGCGGAGAGGGAAGCCGGTGCACAGGTGATGCGTGAGACTGAA CAGCAGATTTACCGTCAGCTGACTGACGAGGTACTGGCCCTGCGATTGCCT GAAAACGGCTCACAACTGCACCATTCATAA MIKSTNIQAIGSGIMHQINNIYSLTPFPLPMELTPSCNEFYLKAWSEWEKN GTPGEQRNIAFNRLKICLQNQEAELNLSELDLKTLPDLPPQITTLEIRKNL LTHLPDLPPMLKVIHAQFNQLESLPALPETLEELNAGDNKIKELPFLPENL THLRVHNNRLHILPLLPPELKLLVVSGNRLDSIPPFPDKLEGLAMANNFIE QLPELPFSMNRAVLMNNNLTTLPESVLRLAQNAFVNVAGNPLSGHTMRTLQ QITTGPDYSGPRIFFSMGNSATISAPEHSLADAVTAWFPENKQSDVSQIWH AFEHEEHANTFAFLDRLSDTVSARNTSGFREQVAAWLEKLSASAELRQQSF AVAADATESCEDRVALTWNNLRKTLLVHQASEGLFDNDTGALLSLGREMFR LEILEDIARDKVRTLHFVDEIEVYLAFQTMLAEKLQLSTAVKEMRFYGVSG VTANDLRTAEAMVRSREENEFKDWFSLWGPWHAVLKRTEADRWAQAEEQKY EMLENEYSQRVADRLKASGLSGDTDAEREAGAQVMRETEQQIYRQLTDEVL ALRLSENGSNHIA ATGATTAAATCAACCAATATACAGGCAATCGGTTCTGGTATTATGCATCAA ATAAACAATATATACTCGTTAACTCCATTTCCTTTACCTATGGAACTGACT CCATCTTGTAATGAATTTTATTTAAAAGCCTGGAGTGAATGGGAAAAGAAC GGTACCCCAGGCGAGCAACGCAATATCGCCTTCAATAGGCTGAAAATATGT TTACAAAATCAAGAGGCAGAATTAAATTTATCTGAGTTAGATTTAAAAACA TTACCAGATTTACCGCCTCAGATAACAACACTGGAAATAAGAAAAAACCTA TTAACACATCTCCCTGATTTACCACCAATGCTTAAGGTAATACATGCTCAA TTTAATCAACTGGAAAGCTTACCTGCCTTACCCGAGACGTTAGAAGAGCTT AATGCGGGTGATAACAAGATAAAAGAATTACCATTTCTTCCTGAAAATCTA ACTCATTTACGGGTTCATAATAACCGATTGCATATTCTGCCACTATTGCCA CCGGAACTAAAATTACTGGTAGTTTCTGGAAACAGATTAGACAGCATTCCC CCCTTTCCAGATAAGCTTGAAGGGCTGGCTATGGCTAATAATTTTATAGAA CAACTACCGGAATTACCTTTTAGTATGAACAGGGCTGTGCTAATGAATAAT AATCTGACAACACTTCCGGAAAGTGTCCTGAGATTAGCTCAGAATGCCTTC GTAAATGTTGCAGGTAATCCACTGTCTGGCCATACCATGCGTACACTACAA CAAATAACCACCGGACCAGATTATTCTGGTCCTCGAATATTTTTCTCTATG GGAAATTCTGCCACAATTTCCGCTCCAGAACACTCCCTGGCTGATGCCGTG ACAGCATGGTTCCCGGAAAACAAACAATCTGATGTATCACAGATATGGCAT GCTTTTGAACATGAAGAGCACGCCAACACCTTTTCCGCGTTCCTTGACCGC CTTTCCGATACCGTCTCTGCACGCAATACCTCCGGATTCCGTGAACAGGTC GCTGCATGGCTGGAAAAACTCAGTGCCTCTGCGGAGCTTCGACAGCAGTCT TTCGCTGTTGCTGCTGATGCCACTGAGAGCTGTGAGGACCGTGTCGCGCTC ACATGGAACAATCTCCGGAAAACCCTCCTGGTCCATCAGGCATCAGAAGGC CTTTTCGATAATGATACCGGCGCTCTGCTCTCCCTGGGCAGGGAAATGTTC CGCCTCGAAATTCTGGAGGACATTGCCCGGGATAAAGTCAGAACTCTCCAT TTTGTGGATGAGATAGAAGTCTACCTGGCCTTCCAGACCATGCTCGCAGAG AAACTTCAGCTCTCCACTGCCGTGAAGGAAATGCGTTTCTATGGCGTGTCG GGAGTGACAGCAAATGACCTCCGCACTGCCGAAGCCATGGTCAGAAGCCGT GAAGAGAATGAATTTAAGGACTGGTTCTCCCTCTGGGGACCATGGCATGCT GTACTGAAGCGTACGGAAGCTGACCGCTGGGCGCAGGCAGAAGAGCAGAAG TATGAGATGCTGGAGAATGAGTACTCTCAGAGGGTGGCTGACCGGCTGAAA GCATCAGGTCTGAGCGGTGATACGGATGCGGAGAGGGAAGCCGGTGCACAG GTGATGCGTGAGACTGAACAGCAGATTTACCGTCAGTTGACTGACGAGGTA CTGGCCCTGCGATTGTCTGAAAACGGCTCAAATCATATCGCATAA MLIRILVIMIKSTNIQAIGSGIMHQINNVYSLTPLSLPMELTPSCNEFYLK TWSEWEKNGTPGEQRNIAFNRLKICLQNQEAELNLSELDLKTLPDLPPQIT TLEIRKNLLTHLPDLPPMLKVIHAQFNQLESLPALPETLEELNAGDNKIKE LPFLPENLTHLRVHNNRLHILPLLPPELKLLVVSGNRLDSIPPFPDKLEGL ALANNFIEQLPELPFSMNRAVLMNNNLTTLPESVLRLAQNAFVNVAGNPLS GHTMRTLQQITTGPDYSGPRIFFSMGNSATISAPEHSLADAVTAWFPENKQ SDVSQIWHAFEHEEHANTFSAFLDRLSDTVSARNTSGFREQVAAWLEKLSA SAELRQQSFAVAADATESCEDRVALTWNNLRKTLLVHQASEGLFDNDTGAL LSLGREMFRLEILEDIARDKVRTLHFVDEIEVYLAFQTMLAEKLQLSTAVK EMRFYGVSGVTANDLRTAEAMVRSREENEFTDWFSLWGPWHAVLKRTEADR WAQAEEQKYEMLENEYSQRVADRLKASGLSGDADAEREAGAQVMRETEQQI YRQLTDEVLA atgTTGATAAGAATTCTAGTTATAATGATTAAATCAACCAATATACAGGCA ATCGGTTCTGGCATTATGCATCAAATAAACAATGTATACTCGTTAACTCCA TTATCTTTACCTATGGAACTGACTCCATCTTGTAATGAATTTTATTTAAAA ACCTGGAGCGAATGGGAAAAGAACGGTACCCCAGGCGAGCAACGCAATATC GCCTTCAATAGGCTGAAAATATGTTTACAAAATCAAGAGGCAGAATTAAAT TTATCTGAGTTAGATTTAAAAACATTACCAGATTTACCGCCTCAGATAACA ACACTGGAAATAAGAAAAAACCTATTAACACATCTCCCTGATTTACCACCA ATGCTTAAGGTAATACATGCTCAATTTAATCAACTGGAAAGCTTACCTGCC TTACCCGAGACGTTAGAAGAGCTTAATGCGGGTGATAACAAGATAAAAGAA TTACCATTTCTTCCTGAAAATCTAACTCATTTACGGGTTCATAATAACCGA TTGCATATTCTGCCACTATTGCCACCGGAACTAAAATTACTGGTAGTTTCT GGAAACAGATTAGACAGCATTCCCCCCTTTCCAGATAAGCTTGAAGGGCTG GCTCTGGCTAATAATTTTATAGAACAACTACCGGAATTACCTTTTAGTATG AACAGGGCTGTGCTAATGAATAATAATCTGACAACACTTCCGGAAAGTGTC CTGAGATTAGCTCAGAATGCCTTCGTAAATGTTGCAGGTAATCCATTGTCT GGCCATACCATGCGTACACTACAACAAATAACCACCGGACCAGATTATTCT GGTCCTCGAATATTTTTCTCTATGGGAAATTCTGCCACAATTTCCGCTCCA GAACACTCCCTGGCTGATGCCGTGACAGCATGGTTCCCGGAAAACAAACAA TCTGATGTATCACAGATATGGCATGCTTTTGAACATGAAGAGCATGCCAAC ACCTTTTCCGCGTTCCTTGACCGCCTTTCCGATACCGTCTCTGCACGCAAT ACCTCCGGATTCCGTGAACAGGTCGCTGCATGGCTGGAAAAACTCAGTGCC TCTGCGGAGCTTCGACAGCAGTCTTTCGCTGTTGCTGCTGATGCCACTGAG AGCTGTGAGGACCGTGTCGCGCTCACATGGAACAATCTCCGGAAAACCCTC CTGGTCCATCAGGCATCAGAAGGCCTTTTCGATAATGATACCGGCGCTCTG CTCTCCCTGGGCAGGGAAATGTTCCGCCTCGAAATTCTGGAGGATATTGCC CGGGATAAAGTCAGAACTCTCCATTTTGTGGATGAGATAGAAGTCTACCTG GCCTTCCAGACCATGCTCGCAGAGAAACTTCAGCTCTCTACTGCCGTGAAG GAAATGCGTTTCTATGGCGTGTCGGGAGTGACAGCAAATGACCTCCGCACT GCCGAAGCCATGGTCAGAAGCCGTGAAGAGAATGAATTTACGGACTGGTTC TCCCTCTGGGGACCATGGCATGCTGTACTGAAGCGTACGGAAGCTGACCGC TGGGCGCAGGCAGAAGAGCAGAAGTATGAGATGCTGGAGAATGAGTACTCT CAGAGGGTGGCTGACCGGCTGAAAGCATCAGGTCTGAGCGGTGATGCGGAT GCGGAGAGGGAAGCCGGTGCACAGGTGATGCGTGAGACTGAACAGCAGATT TACCGTCAGTTGACTGACGAGGTACTGGCCTGA MKPINNHSFFRSLCGLSCISRLSVEEQCTRDYHRIWDDWARE GTTTENRIQAVRLLKICLDTREPVLNLSLLKLRSLPPLPLHI RELNISNNELISLPENSPLLTELHVNGNNLNILPTLPSQLIK LNISFNRNLSCLPSLPPYLQSLSARFNSLETLPELPSTLTIL RIEGNRLTVLPELPHRLQELFVSGNRLQELPEFPQSLKYLKV GENQLRRLSRLPQELLALDVSNNLLTSLPENIITLPICTNVN ISGNPLSTRVLQSLQRLTSSPDYHGPQIYFSMSDGQQNTLII RPLADAVTAWFPENKQSDVSQIWHAFEHEEHANTFSAFLDRL SDTVSARNTSGFREQVAAWLEKLSASAELRQQSFAVAADATE SCEDRVALTWNNLRKTLLVHQASEGLFDNDTGALLSLGREMF RLEILEDIARDKVRTLHFVDEIEVYLAFQTMLAEKLQLSTAV KEMRFYGVSGVTANDLRTAEAMVRSREENEFTDWFSLWGPWH AVLKRTEADRWAQAEEQKYEMLENEYSQRVADRLKASGLSGD ADAQREAGAQVMRETEQQIYRQLTDEVLA ATGAAACCGATCAACAATCATTCTTTTTTTCGTTCCCTTTGT GGCTTATCATGTATATCTCGTTTATCGGTAGAAGAACAGTGT ACCAGAGATTACCACCGCATCTGGGATGACTGGGCTAGGGAA GGAACAACAACAGAAAATCGCATCCAGGCGGTTCGATTATTG AAAATATGTCTGGATACCCGGGAGCCTGTTCTCAATTTAAGC TTACTGAAACTACGTTCTTTACCACCACTCCCTTTGCATATA CGTGAACTTAATATTTCCAACAATGAGTTAATCTCCCTACCT GAAAATTCTCCGCTTTTGACAGAACTTCATGTAAATGGTAAC AACTTGAATATACTCCCGACACTTCCATCTCAACTGATTAAG CTTAATATTTCATTCAATCGAAATTTGTCATGTCTGCCATCA TTACCACCATATTTACAATCACTCTCGGCACGTTTTAATAGT CTGGAGACGTTACCAGAGCTTCCATCAACGCTAACAATATTA CGTATTGAAGGTAATCGCCTTACTGTCTTGCCTGAATTGCCT CATAGACTACAAGAACTCTTTGTTTCCGGCAACAGACTACAG GAACTACCAGAATTTCCTCAGAGCTTAAAATATTTGAAGGTA GGTGAAAATCAACTACGCAGATTATCCAGATTACCGCAAGAA CTATTGGCTCTGGATGTTTCCAATAACCTACTAACTTCATTA CCCGAAAATATAATCACATTGCCCATTTGTACGAATGTTAAC ATTTCAGGGAATCCATTGTCGACTCGCGTTCTGCAATCCCTG CAAAGATTAACCTCTTCGCCGGACTACCACGGCCCGCAGATT TACTTCTCCATGAGTGACGGACAACAGAATACACTCCATCGC CCCCTGGCTGATGCCGTGACAGCATGGTTCCCGGAAAACAAA CAATCTGATGTATCACAGATATGGCATGCTTTTGAACATGAA GAGCATGCCAACACCTTTTCCGCGTTCCTTGACCGCCTTTCC GATACCGTCTCTGCACGCAATACCTCCGGATTCCGTGAACAG GTCGCTGCATGGCTGGAAAAACTCAGTGCCTCTGCGGAGCTT CGACAGCAGTCTTTCGCTGTTGCTGCTGATGCCACTGAGAGC TGTGAGGACCGTGTCGCGCTCACATGGAACAATCTCCGGAAA ACCCTCCTGGTCCATCAGGCATCAGAAGGCCTTTTCGATAAT GATACCGGCGCTCTGCTCTCCCTGGGCAGGGAAATGTTCCGC CTCGAAATTCTGGAGGACATTGCCCGGGATAAAGTCAGAACT CTCCATTTTGTGGATGAGATAGAAGTCTACCTGGCCTTCCAG ACCATGCTCGCAGAGAAACTTCAGCTCTCCACTGCCGTGAAG GAAATGCGTTTCTATGGCGTGTCGGGAGTGACAGCAAATGAC CTCCGCACTGCCGAAGCTATGGTCAGAAGCCGTGAAGAGAAT GAATTTACGGACTGGTTCTCCCTCTGGGGACCATGGCATGCT GTACTGAAGCGTACGGAAGCTGACCGCTGGGCGCAGGCAGAA GAGCAGAAGTATGAGATGCTGGAGAATGAGTACTCTCAGAGG GTGGCTGACCGGCTGAAAGCATCAGGTCTGAGCGGTGATGCG GATGCGCAGAGGGAAGCCGGTGCACAGGTGATGCGTGAGACT GAACAGCAGATTTACCGTCAGCTGACTGACGAGGTACTGGCC TGA MFSVNNTHSSVSCSPSINSNSTSNEHYLRILTEWEKNSSPGE ERGIAFNRLSQCFQNQEAVLNLSDLNLTSLPELPKHISALIV ENNKLTSLPKLPAFLKELNADNNRLSVIPELPESLTTLSVRS NQLENLPVLPNHLTSLFVENNRLYNLPALPEKLKFLHVYYNR LTTLPDLPDKLEILCAQRNNLVTFPQFSDRNNIRQKEYYFHF NQITTLPESFSQLDSSYRINISGNPLSTRVLQSLQRLTSSPD YHGPQIYFSMSDGQQNTLHRPLADAVTAWFPENKQSDVSQIW HAFEHEEHANTFSAFLDRLSDTVSARNTSGFREQVAAWLEKL SASAELRQQSFAVAADATESCEDRVALTWNNLRKTLLVHQAS EGLFDNDTGALLSLGREMFRLEILEDIARDKVRTLHFVDEIE VYLAFQTMLAEKLQLSTAVKEMRFYGVSGVTANDLRTAEAMV RSREENEFTDWFSLWGPWHAVLKRTEADRWAQAEEQKYEMLE NEYSQRVADRLKASGLSGDADAEREAGAQVMRETEQQIYRQL TDEVLALRLSENGSRLHHS ATGTTCTCTGTAAATAATACACACTCATCAGTTTCTTGCTCC CCCTCTATTAACTCAAACTCAACCAGTAATGAACATTATCTG AGAATCCTGACTGAATGGGAAAAGAACTCTTCTCCCGGGGAA GAGCGAGGCATTGCTTTTAACAGACTCTCCCAGTGCTTTCAG AATCAAGAAGCAGTATTAAATTTATCAGACCTAAATTTGACG TCTCTTCCCGAATTACCAAAGCATATTTCTGCTTTGATTGTA GAAAATAATAAATTAACATCATTGCCAAAGCTGCCTGCATTT CTTAAAGAACTTAATGCTGATAATAACAGGCTTTCTGTGATA CCAGAACTTCCTGAGTCATTAACAACTTTAAGTGTTCGTTCT AATCAACTGGAAAACCTTCCTGTTTTGCCAAACCATTTAACA TCATTATTTGTTGAAAATAACAGGCTATATAACTTACCGGCT CTTCCCGAAAAATTGAAATTTTTACATGTTTATTATAACAGG CTGACAACATTACCCGACTTACCGGATAAACTGGAAATTCTC TGTGCTCAGCGCAATAATCTGGTTACTTTTCCTCAATTTTCT GATAGAAACAATATCAGACAAAAGGAATATTATTTTCATTTT AATCAGATAACCACTCTTCCGGAGAGTTTTTCACAATTAGAT TCAAGTTACAGGATTAATATTTCAGGGAATCCATTGTCGACT CGCGTTCTGCAATCCCTGCAAAGATTAACCTCTTCGCCGGAC TACCACGGCCCACAGATTTACTTCTCCATGAGTGACGGACAA CAGAATACACTCCATCGCCCCCTGGCTGATGCCGTGACAGCA TGGTTCCCGGAAAACAAACAATCTGATGTATCACAGATATGG CATGCTTTTGAACATGAAGAGCATGCCAACACCTTTTCCGCG TTCCTTGACCGCCTTTCCGATACCGTCTCTGCACGCAATACC TCCGGATTCCGTGAACAGGTCGCTGCATGGCTGGAAAAACTC AGTGCCTCTGCGGAGCTTCGACAGCAGTCTTTCGCTGTTGCT GCTGATGCCACTGAGAGCTGTGAGGACCGTGTCGCGCTCACA TGGAACAATCTCCGGAAAACCCTCCTGGTCCATCAGGCATCA GAAGGCCTTTTCGATAATGATACCGGCGCTCTGCTCTCCCTG GGCAGGGAAATGTTCCGCCTCGAAATTCTGGAGGACATTGCC CGGGATAAAGTCAGAACTCTCCATTTTGTGGATGAGATAGAA GTCTACCTGGCCTTCCAGACCATGCTCGCAGAGAAACTTCAG CTCTCTACTGCCGTGAAGGAAATGCGTTTCTATGGCGTGTCG GGAGTGACAGCAAATGACCTCCGCACTGCCGAAGCCATGGTC AGAAGCCGTGAAGAGAATGAATTTACGGACTGGTTCTCCCTC TGGGGACCATGGCATGCTGTACTGAAGCGTACGGAAGCTGAC CGCTGGGCGCAGGCAGAAGAGCAGAAGTATGAGATGCTGGAG AATGAGTACTCTCAGAGGGTGGCTGACCGGCTGAAAGCATCA GGTCTGAGCGGTGATGCGGATGCGGAGAGGGAAGCCGGTGCA CAGGTGATGCGTGAGACTGAACAGCAGATTTACCGTCAGTTG ACTGACGAGGTACTGGCCCTGCGATTGTCTGAAAACGGCTCA CGACTGCACCATTCATAA MLPINNNFSLPQNSFYNTISGTYADYFSAWDKWEKQALPGEE RDEAVSRLKECLINNSDELRLDRLNLSSLPDNLPAQITLLNV SYNQLTNLPELPVTLKKLYSASNKLSELPVLPPALESLQVQH NELENLPALPDSLLTMNISYNEIVSLPSLPQALKNLRATRNF LTELPAFSEGNNPVVREYFFDRNQISHIPESILNLRNECSIH ISDNPLSSHALQALQRLTSSPDYHGPRIYFSMSDGQQNTLHR PLADAVTAWFPENKQSDVSQIWHAFEHEEHANTFSAFLDRLS DTVSARNTSGFREQVAAWLEKLSASAELRQQSFAVAADATES CEDRVALTWNNLRKTLLVHQASEGLFDNDTGALLSLGREMTR LEILEDIARDKVRTLHIFVDEIEVYLAFQTMLAEKLQLSTAV KEMRFYGVSGVTANDLRTAEAMVRSREENEFTDWFSLWGPWH AVLKRTEADRWAQAEEQKYEMLENEYPQRVADRLKASGLSGD ADAEREAGAQVMRETEQQIYRQLTDEVLALRLSENGSQLHHS ATGTTACCGATAAATAATAACTTTTCATTGCCCCAAAATTCT TTTTATAACACTATTTCCGGTACATATGCTGATTACTTTTCA GCATGGGATAAATGGGAAAAACAAGCGCTCCCCGGTGAAGAG CGTGATGAGGCTGTCTCCCGACTTAAAGAATGTCTTATCAAT AATTCCGATGAACTTCGACTGGACCGTTTAAATCTGTCCTCG CTACCTGACAACTTACCAGCTCAGATAACGCTGCTCAATGTA TCATATAATCAATTAACTAACCTACCTGAACTGCCTGTTACG CTAAAAAAATTATATTCCGCCAGCAATAAATTATCAGAATTG CCCGTGCTACCTCCTGCGCTGGAGTCACTTCAGGTACAACAC AATGAGCTGGAAAACCTGCCAGCTTTACCCGATTCGTTATTG ACTATGAATATCAGCTATAACGAAATAGTCTCCTTACCATCG CTCCCACAGGCTCTTAAAAATCTCAGAGCGACCCGTAATTTC CTCACTGAGCTACCAGCATTTTCTGAGGGAAATAATCCCGTT GTCAGAGAGTATTTTTTTGATAGAAATCAGATAAGTCATATC CCGGAAAGCATTCTTAATCTGAGGAATGAATGTTCAATACAT ATTAGTGATAACCCATTATCATCCCATGCTCTGCAAGCCCTG CAAAGATTAACCTCTTCGCCGGACTACCACGGCCCACGGATT TACTTCTCCATGAGTGACGGACAACAGAATACACTCCATCGC CCCCTGGCTGATGCCGTGACAGCATGGTTCCCGGAAAACAAA CAATCTGATGTATCACAGATATGGCATGCTTTTGAACATGAA GAGCATGCCAACACCTTTTCCGCGTTCCTTGACCGCCTTTCC GATACCGTCTCTGCACGCAATACCTCCGGATTCCGTGAACAG GTCGCTGCATGGCTGGAAAAACTCAGTGCCTCTGCGGAGCTT CGACAGCAGTCTTTCGCTGTTGCTGCTGATGCCACTGAGAGC TGTGAGGACCGTGTCGCGCTCACATGGAACAATCTCCGGAAA ACCCTCCTGGTCCATCAGGCATCAGAAGGCCTTTTCGATAAT GATACCGGCGCTCTGCTCTCCCTGGGCAGGGAAATGTTCCGC CTCGAAATTCTGGAGGATATTGCCCGGGATAAAGTCAGAACT CTCCATTTTGTGGATGAGATAGAAGTCTACCTGGCCTTCCAG ACCATGCTCGCAGAGAAACTTCAGCTCTCCACTGCCGTGAAG GAAATGCGTTTCTATGGCGTGTCGGGAGTGACAGCAAATGAC CTCCGCACTGCCGAAGCCATGGTCAGAAGCCGTGAAGAGAAT GAATTTACGGACTGGTTCTCCCTCTGGGGACCATGGCATGCT GTACTGAAGCGTACGGAAGCTGACCGCTGGGCGCAGGCAGAA GAGCAGAAATATGAGATGCTGGAGAATGAGTACCCTCAGAGG GTGGCTGACCGGCTGAAAGCATCAGGTCTGAGCGGTGATGCG GATGCGGAGAGGGAAGCCGGTGCACAGGTGATGCGTGAGACT GAACAGCAGATTTACCGTCAGCTGACTGACGAGGTACTGGCC CTGCGATTGTCTGAAAACGGCTCACAACTGCACCATTCATAA MKKNFFSDLPEETIVNTLSFLKANTLARIAQTCQFFNRLAND KHLELHQLRQQHIKRELWGNLMVAARSNNLEEVKKILKKGID PTQTNSYHLNRTPLLAAIEGKAYQTANYLWRKYTFDPNFKDN YGDSPISLLKKQLANPAFKDKEKKQIRALIRGMQEEKIAQSK CLVC ATGAAAAAGAATTTTTTTTCTGATCTTCCTGAGGAAACAATT GTCAATACATTGAGTTTCTTAAAAGCAAACACACTAGCTCGT ATAGCTCAGACATGTCAATTTTTTAATCGCTTGGCTAATGAT AAACATCTGGAGCTGCATCAACTAAGACAACAGCATATAAAG CGAGAGCTATGGGGAAATCTTATGGTGGCGGCAAGAAGCAAT AACCTGGAAGAGGTCAAAAAGATTCTAAAAAAAGGAATCGAT CCAACCCAGACCAATAGCTACCACTTAAATAGAACGCCTTTA CTTGCAGCTATCGAAGGAAAAGCATATCAAACTGCAAATTAC CTCTGGAGAAAATACACTTTCGATCCCAATTTTAAAGATAAC TATGGTGATTCACCTATCTCTCTTCTTAAAAAGCAACTGGCA AATCCAGCCTTCAAGGATAAGGAAAAAAAACAAATACGCGCC TTAATTAGGGGAATGCAAGAAGAAAAAATAGCACAGAGCAAG TGCCTTGTTTGTTAA MKAKYDPTKPGLQKLPPEIKVMILEFLDAKSKLALSQTNYGW RDLILDRPEYTKEITNTLFRLDKKRHRQAIAQMMSGRVTASS MAKLFEELLCFSIPSSYVFLIFFASQKSVALIEVLTVILVFA AITSLAHDLVDYFIESDTKAEKQHAHRRAFQFFAQPSQSAAQ QNLEEENLSADPKACQCEPL ATGAAAGCAAAATACGACCCCACAAAGCCTGGACTCCAAAAG TTACCTCCTGAAATCAAGGTAATGATTCTTGAGTTTCTTGAT GCCAAATCAAAACTAGCTCTTTCACAGACAAATTATGGTTGG CGTGATTTAATTCTAGACCGGCCAGAATATACCAAAGAAATA ACGAATACATTATTTCGTCTTGATAAAAAACGCCATCGTCAA GCAATAGCACAAATGATGTCAGGAAGAGTTACAGCAAGTTCT ATGGCTAAGCTATTTGAAGAATTACTATGTTTTAGCATACCT TCGTCCTATGTGTTTTTAATCTTTTTCGCATCGCAAAAATCT GTGGCGCTTATAGAAGTCTTAACCGTAATCCTTGTGTTTGCT GCAATAACCTCTCTCGCCCATGATCTGGTGGATTATTTTATT GAAAGTGATACAAAAGCTGAGAAACAGCATGCACATCGCCGT GCTTTTCAATTCTTTGCCCAACCCAGTCAAAGCGCTGCACAA CAAAACTTGGAGGAAGAGAATTTAAGTGCTGATCCCAAGGCC TGCCAATGTGAGCCATTGTAG MATRNPFDIDEIKSKYLREAALEANLSHIPETTPTMLTCPID SGFLKDPVITPEGFVYNKSSILKWLETKKEDPQSRKPLTAKD LQPFPELLIIVNRFVETQTNYEKLKNRLVQNARVAARQKEYT EIPDIFLCPISKTLIKTPVITAQGKVYDQEALSNFLIATGNK DETGKKLSIDDVVVFDELYQQIKVYNFYRKREVQKNQIQPSV SNGFGFFSLNFLTSWLWGTEEKKEKTSSDMTY ATGGCGACGCGAAATCCTTTTGATATTGATCATAAAAGTAAA TACTTAAGAGAAGCAGCATTAGAAGCCAATTTATCTCATCCA GAAACAACACCAACAATGCTGACTTGCCCTATTGACAGCGGA TTTCTAAAAGATCCCGTGATCACACCTGAAGGTTTTGTTTAT AATAAATCCTCTATTTTAAAATGGTTAGAAACGAAAAAAGAA GACCCACAAAGCCGTAAACCCTTAACGGCTAAAGATTTGCAA CCATTCCCCGAGTTATTGATTATAGTCAATAGATTTGTTGAG ACACAAACGAACTATGAAAAATTAAAAAACAGATTAGTGCAA AATGCTCGGGTTGCTGCACGCCAAAAAGAATACACTGAAATT CCGGATATATTTCTTTGCCCAATAAGTAAAACGCTTATCAAA ACACCTGTCATTACTGCCCAAGGGAAAGTATATGATCAAGAA GCATTAAGTAACTTTCTTATCGCAACGGGTAATAAAGATGAA ACAGGCAAAAAATTATCCATTGATGATGTAGTGGTGTTTGAT GAACTCTATCAACAGATAAAAGTTTATAATTTTTACCGCAAA CGCGAAGTGCAAAAAAATCAAATTCAACCTTCAGTAAGTAAT GGTTTTGGCTTTTTTAGCTTGAATTTTCTCACCTCATGGTTA TGGGGAACTGAGGAGAAAAAAGAAAAGACATCATCTGATATG ACGTACTAA MPLTSDIRSHSFNLGVEVVRARIVANGRGDITVGGETVSIVY DSTNGRFSSSGGNGGLLSELLLLGFNSGPRALGERMLSMLSD SGEAQSQESIQNKISQCKFSVCPERLQCPLEAIQCPITLEQP EKGIFVKNSDGSDVCTLFDAAAFSRLVGEGLPHPLTREPITA SIIVKHEECIYDDTRGNFIIKGN ATGCCATTAACCTCAGATATTAGATCACATTCATTTAATCTT GGGGTGGAGGTTGTTCGTGCCCGAATTGTAGCCAATGGGCGC GGAGATATTACAGTCGGTGGTGAAACTGTCAGTATTGTGTAT GATTCTACTAATGGGCGCTTTTCATCCAGTGGCGGTAATGGC GGATTGCTTTCTGAGTTATTGCTTTTGGGATTTAATAGTGGT CCTCGAGCCCTTGGTGAGAGAATGCTAAGTATGCTTTCGGAC TCAGGTGAAGCACAATCGCAAGAGAGTATTCAGAACAAAATA TCTCAATGTAAGTTTTCTGTTTGTCCAGAGAGACTTCAGTGC CCGCTTGAGGCTATTCAGTGTCCAATTACACTGGAGCAGCCT GAAAAAGGTATTTTTGTGAAGAATTCAGATGGTTCAGATGTA TGTACTTTATTTGATGCCGCTGCATTTTCTCGTTTGGTTGGT GAAGGCTTACCCCACCCACTGACCCGGGAACCAATAACGGCA TCAATAATTGTAAAACATGAAGAATGCATTTATGACGATACC AGAGGAAACTTCATTATAAAGGGTAATTGA MPVDLTPYILPGVSFLSDIPQETLSEIRNQTIRGEAQIRLGE LMVSIRPMQVNGYFMGSLNQDGLSNDNIQIGLQYIEHIERTL NHGSLTSREVTVLREIEMLENMDLLSNYQLEELLDKIEVCAF NVEHAQLQVPESLRTCPVTLCEPEDGVFMRNSMNSNVCMLYD KMALIHLVKTRAAHPLSRESIAVSMIVGRDNCAFDPDRGNFV LKN ATGCCTGTAGATTTAACGCCTTATATTTTACCTGGGGTTAGTTTTTTGTC TGACATTCCTCAAGAAACCTTGTCTGAGATACGTAATCAGACTATTCGTG GAGAAGCTCAAATAAGACTGGGTGAGTTGATGGTGTCAATACGACCTATG CAGGTAAATGGATATTTTATGGGAAGTCTTAACCAGGATGGTTTATCGAA TGATAATATCCAGATTGGCCTTCAATATATAGAACATATTGAACGTACAC TTAATCATGGTAGTTTGACAAGCCGTGAAGTTACAGTACTGCGTGAAATT GAGATGCTCGAAAATATGGATTTGCTTTCTAACTACCAGTTAGAGGAGTT GTTAGATAAAATTGAAGTATGTGCATTTAATGTGGAGCATGCACAATTGC AAGTGCCAGAGAGCTTACGAACATGCCCTGTTACATTATGTGAACCAGAA GATGGGGTATTTATGAGGAATTCAATGAATTCAAATGTTTGTATGTTGTA TGATAAAATGGCATTAATACATCTTGTTAAAACAAGGGCGGCTCATCCTT TGAGCAGGGAATCAATCGCAGTTTCAATGATTGTAGGAAGAGATAATTGT GCTTTTGACCCTGACAGAGGTAACTTCGTTTTAAAAAATTAA MLPTTNISVNSGVISFESPVDSPSNEDVEVALEKWCAEGEFSENRHEVAS KILDVISTNGETLSISEPITTLPDLLPGSLKELVLNGCTELKSINCLPPN LSSLSMVGCSSLEVINCSIPENVINLSLCHCSSLKHIEGSFPEALRNSVY LNGCNSLNESQCQFLAYDVSQGRACLSKAELTADLIWLSANRTGEESAEE LNYSGCDLSGLSLVGLNLSSVNFSGAVLDDTDLRMSDLSQAVLENCSFKN SILNECNFCYANLSNCIIRALFENSNFSNSNLKNASFKGSSYIQYPPILN EADLTGAIIIPGMVLSGAILGDVKELFSEKSNTINLGGCYIDLSDIQENI LSVLDNYTKSNKSILLTMNTSDDKYNHDKVRAAEELIKKISLDELAAFRP YVKMSLADSFSIHPYLNNANIQQWLEPICDDFFDTIIVISWFNNSIMMYM ENGSLLQAGMYFERHPGAMVSYNSSFIQIVMNGSRRDGMQERFRELYEVY LKNEKVYPVTQQSDFGLCDGSGKPDWDDDSDLAYNWVLLSSQDDGMAMMC SLSHMVDMLSPNTSTNWMSFFLYKDGEVQNTFGYSLSNLFSESFPIFSIP YHKAFSQNFVSGILDILISDNELKERFIEALNSNKSDYKMIADDQQRKLA CVWNPFLDGWELNAQHVDMIMGSHVLKDMPLRKQAEILFCLGGVFCKYSS SDMFGTEYDSPEILRRYANGLIEQAYKTDPQVFGSVYYYNDILDRLQGRN NVFTCTAVLTDMLTEHAKESFPEIFSLYYPVAWR ATGCTGCCCACTACAAATATCTCTGTAAATTCTGGAGTAATATCTTTTGAAAGTCCT GTAGATTCACCATCTAACGAGGATGTTGAAGTTGCCCTCGAAAAGTGGTGTGCTGAG GGAGAATTTAGCGAAAATCGTCATGAGGTTGCATCAAAAATACTTGATGTTATAAGT ACTAATGGAGAGACTTTATCAATCAGTGAGCCAATAACAACATTACCAGACTTGCTT CCAGGTTCTCTGAAAGAACTGGTTTTGAATGGATGTACAGAGCTTAAATCAATAAAC TGCTTACCCCCCAACTTATCTTCATTAAGTATGGTTGGATGCTCATCATTAGAGGTTA TAAATTGCAGCATACCTGAAAATGTCATTAATTTATCTTTATGCCATTGTAGTTCTTT GAAACATATAGAAGGTTCCTTTCCTGAGGCACTCAGAAATTCCGTATATTTAAATGG CTGTAATTCATTAAATGAATCGCAATGTCAATTCCTTGCATATGATGTCAGTCAAGG CCGTGCCTGCCTGAGCAAAGCTGAGCTTACTGCTGACTTAATTTGGTTGTCAGCTAA CCGAACGGGTGAAGAGTCTGCTGAAGAATTGAATTACTCTGGATGTGACTTGTCAG GTCTAAGTCTTGTAGGGCTGAATTTATCATCAGTAAATTTTTCTGGAGCAGTGCTTG ATGATACAGATCTCAGGATGAGTGATTTGTCTCAGGCTGTATTGGAAAACTGTTCTT TTAAAAACTCGATTTTGAATGAATGTAATTTTTGTTATGCTAATTTATCTAATTGTAT TATTAGGGCTTTGTTTGAAAACTCTAATTTCAGCAATTCCAATCTTAAAAATGCATC ATTTAAAGGATCTTCATATATACAATATCCTCCAATTTTGAACGAGGCTGATTTAAC AGGAGCTATTATAATTCCTGGAATGGTTTTAAGTGGTGCTATCTTAGGTGATGTAAA GGAGCTCTTTAGTGAAAAAAGTAATACCATTAATCTAGGAGGGTGTTACATAGATCT ATCTGACATACAGGAAAATATATTATCTGTGTTGGATAACTATACAAAATCAAATAA ATCAATTTTATTGACTATGAATACATCTGATGATAAGTATAACCATGATAAAGTAAG GGCCGCTGAAGAACTTATCAAAAAAATATCTCTTGACGAATTAGCGGCGTTCCGGCC CTATGTTAAGATGTCTTTGGCTGATTCATTTAGTATTCATCCTTATTTGAACAACGCA AATATACAGCAATGGCTCGAGCCTATATGTGATGACTTTTTTGATACTATAATGTCTT GGTTTAATAATTCAATAATGATGTATATGGAGAATGGTAGTTTATTGCAGGCAGGGA TGTATTTTGAGCGACATCCAGGTGCGATGGTATCTTATAATAGTTCCTTTATACAAAT TGTAATGAATGGTTCACGGCGTGATGGAATGCAGGAACGATTTAGGGAACTCTATG AAGTATATTTAAAAAATGAAAAAGTTTATCCTGTCACACAGCAGAGTGATTTTGGAT TGTGCGATGGCTCTGGGAAGCCTGACTGGGATGATGATTCCGATTTGGCTTATAACT GGGTTTTGTTATCATCACAGGATGATGGTATGGCAATGATGTGTTCTTTGAGTCATA TGGTTGATATGTTATCTCCTAATACATCAACTAATTGGATGTCCTTTTTTTTATATAA GGATGGAGAAGTTCAAAATACATTTGGGTATTCATTGAGCAATCTTTTTTCTGAATC ATTTCCAATTTTCAGTATTCCTTATCATAAAGCTTTTTCCCAGAATTTCGTTTCTGGTA TTCTGGATATACTCATTTCTGATAATGAACTCAAAGAGAGATTTATTGAGGCACTTA ATTCCAATAAATCAGATTATAAAATGATTGCTGATGATCAGCAAAGGAAACTTGCCT GTGTCTGGAATCCCTTTCTTGATGGTTGGGAACTGAACGCTCAGCATGTAGATATGA TTATGGGGAGCCATGTATTGAAAGATATGCCACTAAGAAAACAGGCTGAAATATTA TTTTGTTTAGGGGGGGTTTTCTGTAAATACTCATCGAGTGATATGTTTGGTACAGAGT ATGATTCTCCTGAGATTCTACGGAGATATGCAAATGGATTGATTGAACAAGCTTATA AAACAGATCCTCAGGTATTTGGCTCAGTTTATTATTACAATGATATTTTAGACAGGC TACAAGGAAGAAATAATGTTTTTACTTGTACCGCTGTGCTGACTGATATGCTAACGG AGCATGCAAAAGAATCTTTTCCTGAAATATTTTCATTGTATTATCCTGTTGCGTGGCG TTGA MVINMVDVIKFKEPERCDYLYVDENNKVHILLPIVGGDEIGLDNTCQTAV ELITFFYGSAHSGVTKYSAEHQLSEYKRQLEEDIKAINSQKKISPHAYDD LLKEKIERLQQIEKYIELIQVLKKQYDEQNDIRQLRTGGIPQLPSGVKEI IKSSENAFAVRLSPYDNDKFTRFDDPLFNVKRNISKYDTPSRQAPIPIYE GLGYRLRSTLFPEDKTPTPINKKSLRDKVKSTVLSHYKDEDRIDGEKKDE KLNELITNLQNELVKELVKSDPQYSKLSLSKDPRGKEINYDYLVNSLMLV DNDSEIGDWIDTILDATVDSTVWVAQASSPFYDGAKEISSDRDADKISIR VQYLLAEANIYCKTNKLSDANFGEFFDKEPHATEIAKRVKEGFTQGADIE PIIYDYINSNHAELGLKSPLTGKQQQEITDKFTKHYNTIKESPHFDEFFV ADPDKKGNIFSHQGRISCHFLDFFTRQTKGKHPLGDLASHQEALQEGTSN RLHHKNEVVAQGYEKLDQFKKEVVKLLAENKPKELLDYLVATSPTGVPNY SMLSKETQNYIAYNRNWPAIQKELEKATSIPESQKQDLSRLLSRDNLQHD NLSAITWSKYSSKPLLDVELNKIAEGLELTAKIYNEKRGREWWFKGSRNE ARKTQCEELQRVSKEINTLLQSESLTKSQVLEKVLNSIETLDKIDRDISA ESNWFQSTLQKEVRLFRDQLKDICQLDKYAFKSTKLDEIISLEMEEQFQK IQDPAVQQIVRDLPSHCHNDEAIEFFKTLNPEEAAKVASYLSLEYREINK STDKKTLLEQDIPRLFKEVNTQLLSKLKEEKAIDEQVHEKLSQLADKIAP EHFTRNNIIKWSTNPEKLEESNLNEPIKSVQSPTTKQTSKQFREAMGEIT GRNEPPTDTLYTGIIKK ATGGTGATAAACATGGTTGACGTAATCAAATTCAAAGAGCCGGAACGTTGTGATTA TCTATATGTTGATGAAAACAACAAAGTTCATATCCTTTTACCGATTGTAGGAGGAGA TGAAATAGGCCTGGATAATACCTGTCAAACAGCAGTTGAGTTGATCACATTTTTCTA TGGTAGTGCGCACAGTGGTGTGACTAAATATTCTGCTGAACACCAACTCAGTGAATA CAAAAGGCAATTGGAAGAAGACATCAAAGCCATCAATAGTCAAAAGAAAATTTCAC CTCATGCTTATGACGATTTATTAAAAGAGAAAATAGAACGCTTACAGCAAATTGAA AAATACATTGAATTAATTCAAGTACTAAAAAAACAATATGATGAACAAAATGATAT CAGGCAACTTCGTACTGGAGGGATTCCGCAATTACCCTCTGGGGTAAAGGAAATCA TTAAATCCTCTGAAAATGCTTTCGCTGTGAGACTTTCTCCATATGACAACGATAAAT TCACTCGCTTTGATGACCCTTTATTCAATGTCAAAAGAAACATCTCAAAATATGACA CGCCCTCAAGACAAGCTCCTATTCCAATATACGAGGGATTAGGTTATCGCCTGCGTT CAACACTGTTCCCGGAAGATAAAACACCAACTCCAATTAATAAAAAATCACTTAGG GATAAAGTTAAAAGCACTGTTCTTAGTCATTATAAAGATGAAGATAGAATTGATGG AGAAAAAAAAGATGAAAAATTAAACGAACTAATTACTAATCTTCAAAACGAACTTG TAAAAGAGTTAGTAAAAAGTGATCCTCAATATTCGAAACTATCTTTATCTAAAGATC CAAGAGGAAAAGAAATAAATTACGATTATTTAGTAAATAGTTTGATGCTTGTAGAT AACGACTCTGAAATTGGTGATTGGATTGATACTATTCTCGACGCTACAGTAGATTCC ACTGTCTGGGTAGCTCAGGCATCCAGCCCTTTCTATGATGGTGCTAAAGAAATATCA TCAGACCGCGATGCGGACAAGATATCCATCAGAGTTCAGTACCTGTTGGCCGAAGC CAATATTTACTGTAAAACAAACAAATTATCGGATGCTAACTTTGGAGAATTTTTCGA CAAAGAGCCTCATGCTACTGAAATTGCGAAAAGAGTAAAGGAAGGATTTACGCAAG GTGCAGATATAGAACCAATTATATACGACTATATTAACAGCAACCATGCCGAGCTG GGATTAAAATCTCCGTTAACCGGCAAACAACAACAAGAAATCACTGATAAATTTAC AAAACATTATAATACGATTAAAGAATCTCCACATTTTGATGAGTTTTTTGTCGCTGA TCCGGATAAAAAAGGCAATATCTTTTCTCATCAAGGCAGAATCAGTTGTCATTTTCT GGATTTCTTTACTCGACAAACCAAAGGCAAACATCCTCTTGGTGATCTTGCAAGTCA TCAGGAAGCTCTCCAGGAAGGAACCTCCAATCGCTTACATCACAAGAATGAGGTAG TAGCCCAGGGGTACGAAAAACTGGATCAATTCAAGAAAGAGGTTGTCAAACTGCTG GCTGAGAATAAACCAAAAGAATTATTGGATTATTTGGTTGCTACCTCACCTACAGGT GTTCCAAATTACTCCATGCTTTCGAAGGAAACTCAAAATTACATTGCTTATAATCGT AACTGGCCAGCCATTCAAAAAGAGCTGGAAAAGGCTACCAGCATCCCGGAGAGTCA AAAACAAGATCTTTCAAGATTGCTTTCTCGTGATAATTTACAACACGATAATCTAAG CGCAATTACCTGGTCAAAATATTCCTCCAAGCCATTATTGGATGTGGAATTAAATAA AATCGCTGAAGGATTAGAACTCACTGCAAAAATTTACAATGAAAAGAGAGGACGCG AATGGTGGTTTAAAGGTTCAAGAAATGAAGCTCGTAAGACCCAATGTGAAGAATTG CAAAGAGTATCCAAAGAAATCAATACTCTTCTGCAAAGTGAATCTTTAACGAAAAG CCAGGTACTTGAAAAGGTTTTAAATTCTATAGAAACATTAGATAAAATTGACAGAG ACATTTCTGCCGAATCCAATTGGTTTCAAAGTACTCTGCAAAAGGAAGTCAGGTTAT TTCGAGATCAATTGAAAGATATTTGCCAATTGGACAAGTATGCCTTTAAATCAACAA AACTTGATGAAATCATCTCTCTGGAAATGGAAGAACAATTTCAAAAGATACAAGAT CCTGCTGTTCAACAAATTGTCAGGGACTTGCCTTCTCATTGCCACAATGATGAAGCA ATTGAATTCTTTAAGACATTGAACCCTGAAGAGGCAGCAAAAGTAGCTAGCTATTTA AGCCTGGAATACAGGGAAATTAATAAATCAACCGATAAGAAAACTCTCCTAGAACA AGATATTCCCAGACTGTTTAAAGAAGTCAATACGCAGTTACTCTCCAAACTCAAAGA AGAAAAAGCTATTGATGAGCAAGTTCATGAAAAACTCAGTCAACTGGCTGACAAAA TTGCCCCTGAGCATTTTACAAGAAATAACATTATAAAATGGTCTACCAACCCTGAAA AGCTTGAGGAATCAAATCTTAATGAGCCAATCAAATCAGTCCAAAGCCCTACTACTA AACAAACATCAAAACAATTCAGGGAAGCGATGGGTGAAATCACTGGAAGAAATGA GCCTCCTACAGACACTTTGTACACGGGAATTATAAAGAAATAG - MFNITNIQSTARHQSISNEASTEVPLKEEIWNKISAFFSSEHQVEAQNCI AYLCEIPPETASPEEIKSKFECLRMLAFPAYADNIQYSRGGADQYCILSE NSQEILSIVFNTEGYTVEGGGKSVTYTRVTESEQASSASGSKDAVNYELI WSEWVKEAPAKEAANREEPVQRMRDCLKNNKTELRLKILGLTTIPAYIPE QITTLILDNNELKSLPENLQGNIKTLYANSNQLTSIPATLPDTIQEMELS INRITELPERLPSALQSLDLFHNKISCLPENLPEELRYLSVYDNSIRTLP AHLPSEITHLNVQSNSLTALPETLPPGLKTLEAGENALTSLPASLPPELQ VLDVSKNQITVLPETLPPTITTLDVSRNALTNLPENLPAALQIMQASRNN LVRLPESLPHFRGEGPQPTRIIVEYNPFSERTIQNMQRLMSSVDYQGPRV LVAMGDFSIVRVTRPLHQAVQGWLTSLEEEDVNQWRAFEAEANAAAFSGF LDYLGDTQNTRHPDFKEQVSAWLMRLAEDSALRETVFIIAMNATISCEDR VTLAYHQMQEATLVHDAERGAFDSHLAELIMAGREIFRLEQIESLAREKV KRLFFIDEVEVFLGFQNQLRESLSLTTMTRDMRFYNVSGITESDLDEAEI RIKMAENRDFHKWFALWGPWHKVLERIAPEEWREMMAKRDECIETDEYQS RVNAELEDLRIADDSDAERTTEVQMDAERAIGIKIMEEINQTLFTEIMEN ILLKKEVSSLMSAYWR ATGTTTAATATTACTAATATACAATCTACGGCAAGGCATCAAAGTATTAGCAATGAG GCCTCAACAGAGGTGCCTTTAAAAGAAGAGATATGGAATAAAATAAGTGCCTTTTT CTCTTCAGAACATCAGGTTGAAGCACAAAACTGCATCGCTTATCTTTGTCATCCACC TGAAACCGCCTCGCCAGAAGAGATCAAAAGCAAGTTTGAATGTTTAAGGATGTTAG CTTTCCCGGCGTATGCGGATAATATTCAGTATAGTAGAGGAGGGGCAGACCAATAC TGTATTTTGAGTGAAAATAGTCAGGAAATTCTGTCTATAGTTTTTAATACAGAGGGC TATACCGTTGAGGGAGGGGGAAAGTCAGTCACCTATACCCGTGTGACAGAAAGCGA GCAGGCGAGTAGCGCTTCCGGCTCCAAAGATGCTGTGAATTATGAGTTAATCTGGTC TGAGTGGGTAAAAGAGGCGCCAGCGAAAGAGGCAGCAAATCGTGAAGAACCCGTA CAACGGATGCGTGACTGCCTGAAAAATAATAAGACGGAACTTCGTCTGAAAATATT AGGACTTACCACTATACCTGCCTATATTCCTGAGCAGATAACTACTCTGATACTCGA TAACAATGAACTGAAAAGTTTGCCGGAAAATTTACAGGGAAATATAAAGACCCTGT ATGCCAACAGTAATCAGCTAACCAGTATCCCTGCCACGTTACCGGATACCATACAGG AAATGGAGCTGAGCATTAACCGTATTACTGAATTGCCGGAACGTTTGCCTTCAGCGC TTCAATCGCTGGATCTTTTCCATAATAAAATTAGTTGCTTACCTGAAAATCTACCTGA GGAACTTCGGTACCTGAGCGTTTATGATAACAGCATAAGGACACTGCCAGCACATCT TCCGTCAGAGATTACCCATTTGAATGTGCAGAGTAATTCGTTAACCGCTTTGCCTGA AACATTGCCGCCGGGCCTGAAGACTCTGGAGGCCGGCGAAAATGCCTTAACCAGTC TGCCCGCATCGTTACCACCAGAATTACAGGTCCTGGATGTAAGTAAAAATCAGATTA CGGTTCTGCCTGAAACACTTCCTCCCACGATAACAACGCTGGATGTTTCCCGTAACG CATTGACTAATCTACCGGAAAACCTCCCGGCGGCATTACAAATAATGCAGGCCTCTC GCAATAACCTGGTCCGTCTCCCGGAGTCGTTACCCCATTTTCGTGGTGAAGGACCTC AACCTACAAGAATAATCGTAGAATATAATCCTTTTTCAGAACGAACAATACAGAAT ATGCAGCGGCTAATGTCCTCTGTAGATTATCAGGGACCCCGGGTATTGGTTGCCATG GGCGACTTTTCAATTGTTCGGGTAACTCGACCACTGCATCAAGCTGTCCAGGGGTGG CTAACCAGTCTCGAGGAGGAAGACGTCAACCAATGGCGGGCGTTTGAGGCAGAGGC AAACGCGGCGGCTTTCAGCGGATTCCTGGACTATCTTGGTGATACGCAGAATACCCG ACACCCGGATTTTAAGGAACAAGTCTCCGCCTGGCTAATGCGCCTGGCTGAAGATA GCGCACTAAGAGAAACCGTATTTATTATAGCGATGAATGCAACGATAAGCTGTGAA GATCGGGTCACACTGGCATACCACCAAATGCAGGAAGCGACGTTGGTTCATGATGC TGAAAGAGGCGCCTTTGATAGCCACTTAGCGGAACTGATTATGGCGGGGCGTGAAA TCTTTCGGCTGGAGCAAATAGAATCGCTCGCCAGAGAAAAGGTAAAACGGCTGTTT TTTATTGACGAAGTCGAAGTATTTCTGGGGTTTCAGAATCAGTTACGAGAGTCGCTG TCGCTGACAACAATGACCCGGGATATGCGATTTTATAACGTTTCGGGTATCACTGAG TCTGACCTGGACGAGGCGGAAATAAGGATAAAAATGGCTGAAAATAGGGATTTTCA CAAATGGTTTGCGCTGTGGGGGCCGTGGCATAAAGTGCTGGAGCGCATAGCGCCAG AAGAGTGGCGTGAAATGATGGCTAAAAGGGATGAGTGTATTGAAACGGATGAGTAT CAGAGCCGGGTCAATGCTGAACTGGAAGATTTAAGAATAGCAGACGACTCTGACGC AGAGCGTACTACTGAGGTACAGATGGATGCAGAGCGTGCTATTGGGATAAAAATAA TGGAAGAGATCAATCAGACCCTCTTTACTGAGATCATGGAGAATATATTGCTGAAA AAAGAGGTGAGCTCGCTCATGAGCGCCTACTGGCGATAG MKISSGAINFSTIPNQVKKLITSIREHTKNGLTSKITSVKNTHTSLNEKF KTGKDSPIEFALPQKIKDFFQPKDKNTLNKTLITVKNIKDTNNAGKKNIS AEDVSKMNAAFMRKHIANQTCDYNYRMTGAAPLPGGVSVSANNRPTVSEG RTPPVSPSLSLQATSSPSSPADWAKKLTDAVLRQKAGETLTAADRDFSNA DFRNITFSK1LPPSFMERDGDIIKGFNFSNSKFTYSDISHILHFDECRFT YSTLSDVVCSNTKFSNSDMNEVFLQYSITTQQQPSFIDTTLKNTLIRIIK ANLSGVILNEPDNSSPPSVSGGGNFIRLGDIWLQMPLLWTENAVDGFLNH EHNNGKSILMTIDSLPDKYSQEKVQAMEDLVKSLRGGRLTEACIRPVESS LVSVLAHPPYTQSALISEWLGPVQERFFAHQCQTYNDVPLPAPDTYYQQR ILPVLLDSFDRNSAAMTTHSGLFNQVILHCMTGVDCTDGTRQKAAALYEQ YLAHPAVSPHIHNGLFGNYDGSPDWTTRAADNFLLLSSQDSDTAMMLSTD TLLTMLNPTPDTAWDNFYLLRAGENVSTAQISPVELFRHDFPVFLAAFNQ QATQRRFGELIDIILSTEEHGELNQQFLAATNQKHSTVKLIDDASVSRLA TIFDPLLPEGKLSPAHYQHILSAYHLTDATPQKQAETLFCLSTAFARYSS SAIFGTEHDSPPALRGYAEALMQKAWELSPAIFPSSEQFTEWSDRFHGLH GAFTCTSVVADSMQRHARKYFPSVLSSILPLAWA ATGAAGATATCATCAGGCGCAATTAATTTTTCTACTATTCCTAACCAGGTTAAAAAA TTAATTACCTCTATTCGTGAACATACGAAAAACGGGCTCACCTCAAAAATAACCAGT GTTAAAAACACGCATACATCTTTAAATGAAAAATTTAAAACAGGAAAGGACTCACC GATTGAGTTCGCGTTACCACAAAAAATAAAAGACTTCTTTCAGCCGAAAGATAAAA ACACCTTAAACAAAACATTGATTACTGTTAAAAATATTAAAGATACAAATAATGCA GGCAAGAAAAATATTTCAGCAGAAGATGTCTCAAAAATGAATGCAGCATTCATGCG TAAGCATATTGCAAATCAAACATGTGATTATAATTACAGAATGACAGGTGCGGCCC CGCTCCCCGGTGGAGTCTCTGTATCAGCCAATAACAGGCCCACGGTTTCTGAAGGTA GAACACCACCAGTATCCCCCTCCCTCTCACTTCAGGCTACCTCTTCCCCGTCATCACC TGCCGACTGGGCTAAGAAACTCACGGATGCAGTTTTACGACAGAAAGCCGGAGAAA CCCTTACGGCCGCAGATCGCGATTTTTCAAACGCAGATTTCCGTAATATTACATTCA GCAAAATATTGCCCCCCAGCTTCATGGAGCGAGACGGCGATATTATTAAGGGGTTC AACTTTTCAAATTCAAAATTTACTTATTCTGATATATCTCATTTACATTTTGACGAAT GCCGATTCACTTATTCGACACTGAGTGATGTAGTCTGCAGTAATACGAAATTTAGTA ATTCAGACATGAATGAAGTGTTTTTACAGTATTCAATTACTACACAACAACAGCCCT CGTTTATTGATACAACATTAAAAAATACGCTTATACGTCACAAAGCCAACCTCTCTG GCGTTATTTTAAATGAACCGGATAATTCATCACCTCCGTCAGTGTCAGGGGGCGGAA ATTTTATTCGTCTAGGTGATATCTGGCTGCAAATGCCACTCCTTTGGACTGAGAACG CTGTGGATGGATTTTTAAATCATGAGCACAATAATGGTAAAAGTATTCTGATGACCA TTGACAGCCTGCCCGATAAATACAGTCAGGAAAAAGTCCAGGCAATGGAAGACCTG GTTAAGTCATTGCGGGGTGGCCGCTTAACAGAGGCATGTATCCGGCCAGTTGAAAG TTCGCTGGTAAGCGTACTGGCCCACCCCCCCTATACGCAAAGTGCGCTTATCAGCGA GTGGCTCGGGCCTGTTCAGGAACGTTTTTTTGCCCACCAGTGCCAGACCTATAATGA CGTTCCCCTGCCGGCTCCTGACACATATTATCAGCAGCGCATACTGCCTGTGTTGCT GGATTCGTTTGACAGGAACAGCGCCGCCATGACCACTCACAGCGGACTCTTTAATCA GGTGATTTTACACTGTATGACAGGCGTGGACTGCACTGATGGCACCCGCCAGAAAG CTGCAGCGCTTTATGAACAGTATCTTGCTCACCCGGCGGTGTCTCCCCACATCCATA ATGGGCTCTTCGGCAATTATGATGGCAGCCCGGACTGGACAACCCGCGCTGCAGAT AATTTCCTGCTGCTTTCCTCCCAAGATTCAGACACGGCGATGATGCTCTCCACTGAC ACGCTGTTAACAATGCTAAACCCTACTCCTGACACTGCATGGGACAACTTTTACCTG CTGCGAGCCGGAGAGAACGTTTCCACCGCGCAAATCTCTCCGGTAGAGTTATTCCGT CATGACTTTCCGGTGTTTCTCGCCGCATTTAATCAGCAGGCCACGCAGCGACGCTTT GGGGAGCTGATTGATATCATCCTCAGCACTGAAGAGCACGGGGAGCTGAACCAGCA GTTTCTTGCCGCCACGAACCAGAAACATTCCACCGTGAAGTTGATTGATGATGCCTC AGTGTCGCGTCTGGCCACCATTTTTGACCCCTTGCTTCCTGAAGGCAAACTCAGCCC GGCACACTACCAGCACATCCTCAGTGCTTATCACCTGACGGACGCCACCCCACAGA AGCAGGCGGAAACCCTGTTCTGTCTCAGTACCGCATTCGCACGCTATTCCTCCAGCG CCATTTTCGGCACTGAGCATGACTCTCCGCCGGCCCTGAGAGGCTATGCGGAGGCGC TGATGCAGAAAGCCTGGGAGCTGTCTCCGGCGATATTCCCATCCAGCGAACAGTTTA CCGAGTGGTCCGACCGTTTTCACGGCCTCCATGGCGCCTTTACCTGTACCAGCGTTG TGGCGGATAGTATGCAACGTCATGCCAGAAAATATTTCCCGAGTGTTCTGTCATCCA TCCTGCCACTGGCCTGGGCGTAA MFNIRNTQPSVSMQAIAGAAAPEASPEEIVWEKIQVFFPQENYEEAQQCL AELCHPARGMLPDHISSQFARLKALTFPAWEENIQCNRDGINQFCILDAG SKEILSITLDDAGNYTVNCQGYSEAHDFIMDTEPGEECTEFAEGASGTSL RPATTVSQKAAEYDAVWSKWERDAPAGESPGRAAVVQEMRDCLNNGNPVL NVGASGLTTLPDRLPPHITTLVIPDNNLTSLPELPEGLRELEVSGNLQLT SLPSLPQGLQKLWAYNNWLASLPTLPPGLGDLAVSNNQLTSLPEMPPALR ELRVSGNNLTSLPALPSGLQKLWAYNNRLTSLPEMSPGLQELDVSHNQLT RLPQSLTGLSSAARVYLDGNPLSVRTLQALRDIIGHSGIRIHFDMAGPSV PREARALEILAVADWLTSAREGEAAQADRWQAFGLEDNAAAFSLVLDRLR ETENFKKDAGFKAQISSWLTQLAEDAALRAKTFAMATEATSTCEDRVTHA LHQMNNVQLVHNAEKGEYDNNLQGLVSTGREMFRLATLEQIAREKAGTLA LVDDVEVYLAFQNKLKESLELTSVTSEMRFFDVSGVTVSDLQAAELQVKT AENSGFSKWILQWGPLHSVLERKVPERFNALREKQISDYEDTYRKLYDEV LKSSGLVDDTDAERTIGVSAMDSAKKEFLDGLRALVDEVLGSYLTARWRL N ATGTTTAATATCCGCAATACACAACCTTCTGTAAGTATGCAGGCTATTGCTGGTGCA GCGGCACCAGAGGCATCTCCGGAAGAAATTGTATGGGAAAAAATTCAGGTTTTTTTC CCGCAGGAAAATTACGAAGAAGCGCAACAGTGTCTCGCTGAACTTTGCCATCCGGC CCGGGGAATGTTGCCTGATCATATCAGCAGCCAGTTTGCGCGTTTAAAAGCGCTTAC CTTCCCCGCGTGGGAGGAGAATATTCAGTGTAACAGGGATGGTATAAATCAGTTTTG TATTCTGGATGCAGGCAGCAAGGAGATATTGTCAATCACTCTTGATGATGCCGGGAA CTATACCGTGAATTGTCAGGGGTACAGTGAAGCACATGACTTCATCATGGACACAG AACCGGGAGAGGAATGCACAGAATTCGCGGAGGGGGCATCCGGGACATCCCTCCGC CCTGCCACAACGGTTTCACAGAAGGCAGCAGAGTATGATGCTGTCTGGTCAAAATG GGAAAGGGATGCACCAGCAGGAGAGTCACCCGGCCGCGCAGCAGTGGTACAGGAA ATGCGTGATTGCCTGAATAACGGCAATCCAGTGCTTAACGTGGGAGCGTCAGGTCTT ACCACCTTACCAGACCGTTTACCACCGCATATTACAACACTGGTTATTCCTGATAAT AATCTGACCAGCCTGCCGGAGTTGCCGGAAGGACTACGGGAGCTGGAGGTCTCTGG TAACCTACAACTGACCAGCCTGCCATCGCTGCCGCAGGGACTACAGAAGCTGTGGG CCTATAATAATTGGCTGGCCAGCCTGCCGACGTTGCCGCCAGGACTAGGGGATCTGG CGGTCTCTAATAACCAGCTGACCAGCCTGCCGGAGATGCCGCCAGCACTACGGGAG CTGAGGGTCTCTGGTAACAACCTGACCAGCCTGCCGGCGCTGCCGTCAGGACTACA GAAGCTGTGGGCCTATAATAATCGGCTGACCAGCCTGCCGGAGATGTCGCCAGGAC TACAGGAGCTGGATGTCTCTCATAACCAGCTGACCCGCCTGCCGCAAAGCCTCACGG GTCTGTCTTCAGCGGCACGCGTATATCTGGACGGGAATCCACTGTCTGTACGCACTC TGCAGGCTCTGCGGGACATCATTGGCCATTCAGGCATCAGGATACACTTCGATATGG CGGGGCCTTCCGTCCCCCGGGAAGCCCGGGCACTGCACCTGGCGGTCGCTGACTGG CTGACGTCTGCACGGGAGGGGGAAGCGGCCCAGGCAGACAGATGGCAGGCGTTCG GACTGGAAGATAACGCCGCCGCCTTCAGCCTGGTCCTGGACAGACTGCGTGAGACG GAAAACTTCAAAAAAGACGCGGGCTTTAAGGCACAGATATCATCCTGGCTGACACA ACTGGCTGAAGATGCTGCGCTGAGAGCAAAAACCTTTGCCATGGCAACAGAGGCAA CATCAACCTGCGAGGACCGGGTCACACATGCCCTGCACCAGATGAATAACGTACAA CTGGTACATAATGCAGAAAAAGGGGAATACGACAACAATCTCCAGGGGCTGGTTTC CACGGGGCGTGAGATGTTCCGCCTGGCAACACTGGAACAGATTGCCCGGGAAAAAG CCGGAACACTGGCTTTAGTCGATGACGTTGAGGTCTATCTGGCGTTCCAGAATAAGC TGAAGGAATCACTTGAGCTGACCAGCGTGACGTCAGAAATGCGTTTCTTTGACGTTT CCGGCGTGACGGTTTCAGACCTTCAGGCTGCGGAGCTTCAGGTGAAAACCGCTGAA AACAGCGGGTTCAGTAAATGGATACTGCAGTGGGGGCCGTTACACAGCGTGCTGGA ACGCAAAGTGCCGGAACGCTTTAACGCGCTTCGTGAAAAGCAAATATCGGATTATG AAGACACGTACCGGAAGCTGTATGACGAAGTGCTGAAATCGTCCGGGCTGGTCGAC GATACCGATGCAGAACGTACTATCGGAGTAAGTGCGATGGATAGTGCGAAAAAAGA ATTTCTGGATGGCCTGCGCGCTCTTGTGGATGAGGTGCTGGGTAGCTATCTGACAGC CCGGTGGCGTCTTAACTAA MPFHIGSGCLPATISNRRIYRIAWSDTPPEMSSWEKMKEFFCSTHQTEAL ECIWTICIIPPAGTTREDVINRFELLRTLAYAGWEESIHSGQHGENYFCI LDEDSQEILSVTLDDAGNYTVNCQGYSETHRLTLDTAQGEEGTGHAEGAS GTFRTSFLPATTAPQTPAEYDAVWSAWRRAAPAEESRGRAAVVQKMRACL NNGNAVLNVGESGLTTLPDCLPAHITTLVIPDNNLTSLPALPPELRTLEV SGNQLTSLPVLPPGLLELSIFSNPLTHLPALPSGLCKLWIFGNQLTSLPV LPPGLQELSVSDNQLASLPALPSELCKLWAYNNQLTSLPMLPSGLQELSV SDNQLASLPTLPSELYKLWAYNNRLTSLPALPSGLKELIVSGNRLTSLPV LPSELKELMVSGNRLTSLPMLPSGLLSLSVYRNQLTRLPESLIHLSSETT VNLEGNPLSERTLQALREITSAPGYSGPIIRFDMAGASAPRETRALHLAA ADWLVPAREGEPAPADRWHMIFGQEDNADAFSLFLDRLSETENFIKDAGF KAQISSWLAQLAEDEALRANTFAMATEATSSCEDRVTFFLHQMKNVQLVH NAEKGQYDNDLAALVATGREMFRLGKLEQIAREKVRTLALVDEIEVWLAY QNKLKKSLGLTSVTSEMRFFDVSGVTVTDLQDAELQVKAAEKSEFREWIL QWGPLEIRVLERKAPERVNALREKQISDYEETYRMLSDTELRPSGLVGNT DAERTIGARAMESAKKTFLDGLRPLVEEMLGSYLNVQWRRN ATGCCCTTTCATATTGGAAGCGGATGTCTTCCCGCCACCATCAGTAATCGCCGCATT TATCGTATTGCCTGGTCTGATACCCCCCCTGAAATGAGTTCCTGGGAAAAAATGAAG GAATTTTTTTGCTCAACGCACCAGACTGAAGCGCTGGAGTGCATCTGGACGATTTGT CACCCGCCGGCCGGAACGACGCGGGAGGATGTGATCAACAGATTTGAACTGCTCAG GACGCTCGCGTATGCCGGATGGGAGGAAAGCATTCATTCCGGCCAGCACGGGGAAA ATTACTTCTGTATTCTGGATGAAGACAGTCAGGAGATATTGTCAGTCACCCTTGATG ATGCCGGGAACTATACCGTAAATTGCCAGGGGTACAGTGAAACACATCGCCTCACC CTGGACACAGCACAGGGTGAGGAGGGCACAGGACACGCGGAAGGGGCATCCGGGA CATTCAGGACATCCTTCCTCCCTGCCACAACGGCTCCACAGACGCCAGCAGAGTATG ATGCTGTCTGGTCAGCGTGGAGAAGGGCTGCACCCGCAGAAGAGTCACGCGGCCGT GCAGCAGTGGTACAGAAAATGCGTGCCTGCCTGAATAATGGCAATGCAGTGCTTAA CGTGGGAGAATCAGGTCTTACCACCTTGCCAGACTGTTTACCCGCGCATATTACCAC ACTGGTTATTCCTGATAATAATCTGACCAGCCTGCCGGCGCTGCCGCCAGAACTGCG GACGCTGGAGGTCTCTGGTAACCAGCTGACTAGCCTGCCGGTGCTGCCGCCAGGACT ACTGGAACTGTCGATCTTTAGTAACCCGCTGACCCACCTGCCGGCGCTGCCGTCAGG ACTATGTAAGCTGTGGATCTTTGGTAATCAACTGACCAGCCTGCCGGTGTTGCCGCC AGGGCTACAGGAGCTGTCGGTATCTGATAACCAACTGGCCAGCCTGCCGGCGCTGC CGTCAGAATTATGTAAGCTGTGGGCCTATAATAACCAGCTGACCAGCCTGCCGATGT TGCCGTCAGGGCTACAGGAGCTGTCGGTATCTGATAACCAACTGGCCAGCCTGCCG ACGCTGCCGTCAGAATTATATAAGCTGTGGGCCTATAATAATCGGCTGACCAGCCTG CCGGCGTTGCCGTCAGGACTGAAGGAGCTGATTGTATCTGGTAACCGGCTGACCAGT CTGCCGGTGCTGCCGTCAGAACTGAAGGAGCTGATGGTATCTGGTAACCGGCTGAC CAGCCTGCCGATGCTGCCGTCAGGACTACTGTCGCTGTCGGTCTATCGTAACCAGCT GACCCGCCTGCCGGAAAGTCTCATTCATCTGTCTTCAGAGACAACCGTAAATCTGGA AGGGAACCCACTGTCTGAACGTACTTTGCAGGCGCTGCGGGAGATCACCAGCGCGC CTGGCTATTCAGGCCCCATAATACGATTCGATATGGCGGGAGCCTCCGCCCCCCGGG AAACTCGGGCACTGCACCTGGCGGCCGCTGACTGGCTGGTGCCTGCCCGGGAGGGG GAACCGGCTCCTGCAGACAGATGGCATATGTTCGGACAGGAAGATAACGCCGACGC ATTCAGCCTCTTCCTGGACAGACTGAGTGAGACGGAAAACTTCATAAAGGACGCGG GGTTTAAGGCACAGATATCGTCCTGGCTGGCACAACTGGCTGAAGATGAGGCGTTA AGAGCAAACACCTTTGCTATGGCAACAGAGGCAACCTCAAGCTGCGAGGACCGGGT CACATTTTTTTTGCACCAGATGAAGAACGTACAGCTGGTACATAATGCAGAAAAAG GGCAATACGATAACGATCTCGCGGCGCTGGTTGCCACGGGGCGTGAGATGTTCCGT CTGGGAAAACTGGAACAGATTGCCCGGGAAAAGGTCAGAACGCTGGCTCTCGTTGA TGAAATTGAGGTCTGGCTGGCGTATCAGAATAAGCTGAAGAAATCACTCGGGCTGA CCAGCGTGACGTCAGAAATGCGTTTCTTTGACGTATCCGGCGTGACGGTTACAGACC TTCAGGACGCGGAGCTTCAGGTGAAAGCCGCTGAAAAAAGCGAGTTCAGGGAGTGG ATACTGCAGTGGGGGCCGTTACACAGAGTGCTGGAGCGCAAAGCGCCGGAACGCGT TAACGCGCTTCGTGAAAAGCAAATATCGGATTATGAGGAAACGTACCGGATGCTGT CTGACACAGAGCTGAGACCGTCTGGGCTGGTCGGTAATACCGATGCAGAGCGCACT ATCGGAGCAAGAGCGATGGAGAGCGCGAAAAAGACATTTTTGGATGGCCTGCGACC TCTTGTGGAGGAGATGCTGGGGAGCTATCTGAACGTTCAGTGGCGTCGTAACTGA MRRVDQPRPPGTPFGLREQTTSNADAPARTAPPAHPAPERPTGMLGGLTR YVPGDRSGRPPAMPAAAETSRRPTTSARPLPYGGSGSAARMNEAAGHPLR MPQLPQLSDIERARFHSVTTDSQHLRPVRPRMPPPVGASPLRRSTALRPY HDVLSQWQRHYNADRNRWHSAWRQANSNNPQIETRTGRALKATADLLEDA TQPGRVALELRSVPLPQFPDQAFRLSHLQHMTIDAAGLMELPDTMQQFAG LETLTLARNPLRALPASIASLNRLRELSIRACPELTELPEPLASTDASGE HQGLVNLQSLRLEWTGIRSLPASIANLQNLKSLKIRNSPLSALGPAIHHL PKLEELDLRGCTALRNYPPIFGGRAPLKRLILKDCSNLLTLPLDIHRLTQ LEKLDLRGCVNLSRLPSLIAQLPANCIILVPPHLQAQLDQHRPVARPAEP GRTGPTTPALSPSAAGDRAGPSSSATASELLLTAALERIEDTAQAMLSTV IDEERNPFLEGAPSYLPGKRPTDVTTFGQVPALRDMLAESRDLEFLQRVS DMAGPSPRIEDPSEEGLARHYTNVSNWKAQKSAHLGIVDHLGQFVYHEGS PLDVATLAKAVQMWKTRELIVHAHPQDRARFPELAVHIPEQVSDDSDSEQ QTSPEPSGHQ ATGCGACGCGTCGATCAACCACGCCCGCCGGGCACGCCTTTCGGACTGCGGGAGCA GACTACGTCCAATGCGGATGCGCCCGCGCGCACTGCCCCACCCGCACACCCCGCGC CCGAGCGCCCTACCGGCATGCTCGGCGGACTGACCAGATATGTGCCTGGCGATCGG TCCGGGCGACCGCCAGCAATGCCTGCCGCTGCCGAGACCTCTCGCCGGCCAACCAC CTCCGCCCGCCCGCTTCCCTACGGCGGATCCGGCAGCGCCGCGCGGATGAACGAGG CGGCTGGACATCCTTTGCGGATGCCGCAATTGCCACAGCTCAGCGACATAGAACGC GCTCGCTTCCACTCCGTCACCACCGACTCGCAACACTTGCGGCCGGTGCGCCCCCGT ATGCCACCGCCCGTGGGCGCTTCACCCTTACGGCGCTCCACAGCGCTGCGCCCGTAC CACGACGTGCTGTCGCAATGGCAACGCCACTACAACGCAGATCGCAATCGCTGGCA CAGCGCATGGCGCCAGGCCAACAGCAACAACCCGCAGATCGAGACTCGCACAGGCC GGGCGCTGAAGGCGACAGCCGACCTGCTGGAGGACGCAACCCAACCGGGCCGGGTC GCGCTGGAGCTGCGCTCAGTTCCGCTGCCGCAATTTCCCGACCAGGCATTCCGTCTT TCGCATCTGCAGCACATGACGATCGACGCGGCAGGGTTGATGGAGCTCCCGGACAC CATGCAGCAATTTGCGGGCCTGGAAACACTCACGCTCGCACGCAATCCGCTTCGCGC GCTACCGGCATCCATCGCAAGCCTCAACCGATTACGCGAGCTCTCCATCCGCGCCTG CCCGGAATTGACGGAACTTCCCGAACCCCTGGCAAGCACCGATGCATCCGGCGAGC ACCAGGGCTTGGTCAACCTGCAGAGCCTACGGCTGGAATGGACCGGGATCAGATCG CTTCCGGCGTCCATCGCCAACCTGCAAAATCTGAAAAGCCTGAAGATACGCAACTC GCCGCTGTCCGCCCTTGGCCCGGCCATCCATCACCTGCCAAAGTTGGAGGAGCTTGA TTTGCGGGGCTGTACCGCGCTGCGCAACTATCCGCCGATTTTCGGCGGCCGTGCGCC ACTGAAGCGACTGATTCTGAAAGACTGCAGCAACCTGCTCACGCTGCCACTGGACA TTCACCGCCTGACGCAGCTGGAAAAACTCGATCTGCGAGGTTGCGTCAACCTTTCCA GACTGCCCTCGTTGATCGCCCAATTACCTGCCAATTGCATCATCCTGGTGCCGCCGC ATCTCCAAGCGCAGCTCGACCAGCATCGTCCAGTTGCGCGCCCCGCCGAACCAGGG CGGACCGGACCGACCACCCCAGCTCTCTCGCCCTCTGCTGCCGGCGACCGCGCCGGG CCATCCTCTTCGGCGACCGCCAGCGAACTGCTTCTTACCGCTGCGCTCGAACGCATC GAAGACACCGCACAGGCCATGCTGAGCACGGTCATCGATGAAGAAAGAAATCCCTT TCTGGAAGGTGCTCCATCCTATCTCCCAGGAAAACGCCCTACCGATGTCACCACCTT CGGCCAAGTTCCGGCATTGCGGGACATGCTGGCAGAAAGCAGGGATCTTGAGTTCC TGCAACGGGTAAGCGACATGGCAGGCCCATCCCCCAGAATCGAAGACCCGAGCGAG GAAGGCCTCGCCCGCCACTACACGAACGTCAGCAACTGGAAGGCGCAGAAGAGCGC ACACCTGGGCATCGTCGATCATCTCGGGCAGTTCGTTTATCACGAAGGAAGCCCGCT CGACGTAGCGACATTGGCCAAGGCAGTGCAGATGTGGAAGACCCGTGAGCTGATCG TCCACGCACACCCGCAAGACCGCGCGCGCTTTCCCGAGCTCGCTGTGCACATTCCCG AGCAGGTCAGCGACGACTCTGATAGCGAACAGCAGACAAGCCCGGAACCTTCAGGC CATCAGTAG EXAMPLES
Experimental Methods
Bacterial strain DH5α F- (Φ80Δ/acZΔM15,) Δ(lac/ZYA-argF)U169 Laboratory stock recA1 endA1 hsdR17(rk supE44 λ- thi-1 gyrA96 relA1 BL21 (DE3) F- ompT gal dcm lon hsdSB(rB Laboratory stock Rosetta(DE3) F- ompT gal dcm lon hsdSB(rB Laboratory stock pRARE (CmR) Cell line HEK293T Laboratory stock HEK293T-EGFP HEK293T cells stably expressing EGFP This study HEK293T-ERK2-EGFP HEK293T cells stably expressing ERK2-EGFP This study HEK293T-EGFP-HRasG12V HEK293T cells stably expressing EGFP-HRasG12V This study HEK293T-d2EGFP HEK293T cells stably expressing d2EGFP This study HeLa H2B-EGFP HeLa cells stably expressing H2B-EGFP fusion [1] MCF-10A Laboratory stock MCF-10A rtTA Laboratory stock MCF-10A rtTA EGFP- MCF-10A cells stably expressing EGFP- This study HRASG12V HRASG12Vfusion and rtTA MCF-10A rtTA GS2-IpaH9.8 MCF-10A cells stably expressing GS2- This study IpaH9.8 fusion and rtTA MCF-10A rtTA EGFP- MCF-10A cells stably expressing EGFP- This study HRASG12V+ GS2-IpaH9.8 HRASG12Vfusion, GS2-IpaH9.8, and rtTA MCF-10A rtTA EGFP MCF-10A cells stably expressing EGFP- This study HRASG12V+ GS2-IpaH9.8C337A HRASG12Vfusion, GS2-IpaH9.8C337A, and rtTA Plasmid Relevant features Source pcDNA3 CMV promoter, AmpR Laboratory stock pCDH1-CMV-MCS-EF1α- CMV promoter; PurR, AmpR Laboratory stock Puro pET28a(+) T7lac promoter; KanR Novagen pET24d(+) T7lac promoter; KanR Novagen pTriEx-3 CMV promoter; T7 promoter; AmpR Novagen psPAX2 Lentiviral packaging vector; CMV promoter; AmpR Laboratory stock pMD2.G Lentiviral packaging vector; CMV promoter; AmpR Laboratory stock pPB TetOn Hygro Transposon vector; SV40 promoter; AmpR Laboratory stock pLV rtTA-NeoR Tetracycline inducible reverse transcriptional Laboratory stock transactivator; CMV promoter; EF1α promoter; NeoR; AmpR pCMV-hyPBase PiggyBac transposase; CMV promoter; AmpR [2] pHIV-d2EGFP Lentiviral vector expressing d2EGFP EF-1α [3] promoter, IRES; AmpR pGEM4Z/GFP/A64 In vitro transcription of GFP with 3′ 64 residue [4] poly A tail; T7 promoter; AmpR pHFT2-GS2 GS2 monobody in pHFT2 plasmid; T7 promoter, [5] KanR pHFT2-GS5 GS5 monobody in pHFT2 plasmid; T7 promoter, [5] KanR pHFT2-GL4 GL4 monobody in pHFT2 plasmid; T7 promoter, [5] KanR PHFT2-GL6 GL6 monobody in pHFT2 plasmid; T7 promoter, [5] KanR pGalAga-GL8 GL8 monobody in pGalAgaCamR; CamR [5] pHFT2-AblSH2MB#AS15 AS15 monobody in pHFT2 plasmid; T7 [4] pHFT2-SHP2NSa5 NSa5 monobody in pHFT2 plasmid; T7 [6] pET24a(+)-RasInll RasInll with C-terminal GS linker, 6xHis, Avi, [7] and Flag tags; T7lac promoter; KanR pcDNA3-R4-CHIPΔTPR scFv13-R4 fused to CHIP lacking TPR [8] domain with C- terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; pcDNA3_NSlmb-vhhGFP4 vhhGFP4 fused to F-box domain from [1]; Addgene #35579 pETHis6MEK1 R4F + ERK2 pET-based expression of two proteins: [9] constitutively active MEK1 and wild-type, pHFT2-SHP2 Full-length SHP2 in pHFT2 plasmid; T7 [6] pcDNA3-EGFP EGFP cloned in pcDNA3; CMV promoter, AmpR; This study pcDNA3-HF-GS2 GS2 with N-terminal Kozak, Flag and 6xHis This study tags,and BamHI and EcoRI sites; CMV pcDNA3-GS2-FH GS2 with C-terminal Nhel and Sbfl sites, Flag This study tag, and 6xHis tag; CMV promoter; AmpR. pcDNA3-GS2-AvrPtoB GS2 fused to This study lacking N- terminal domain with C-terminal Flag and 6x-His tags cloned in pcDNA3; CMV pcDNA3-GS2-IpaH9.8 GS2 fused to This study LRR domain (IpaH9.8ΔLRR) with C-terminal Flag and 6x-His tags cloned in pcDNA3; CMV pcDNA3-GS2-IpaH9.8C337A pcDNA3-GS2-IpaH9.8 containing Cys337Ala This study pcDNA3-AS15-IpaH9.8 AS15 fused to IpaH9.8 lacking LRR domain This study with C-terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; AmpR pcDNA3-GS2-IpaH1.4 GS2 fused to This study domain with C- terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; pcDNA3-GS2-IpaH2.5 GS2 fused to This study domain with C- terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; pcDNA3-GS2-IpaH4.5 GS2 fused to This study domain with C- terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; pcDNA3-GS2-IpaH7.8 GS2 fused to This study domain with C- terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; pcDNA3-GS2-IpaH0722 GS2 fused to This study LRR domain with C-terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; pcDNA3-LegAU13-GS2 GS2 fused to This study with N-terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; AmpR pcDNA3-LegU1-GS2 GS2 fused to This study with N-terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; AmpR pcDNA3-LubX-GS2 GS2 fused to This study LubX lacking CTD domain with N-terminal Flag and 6x-His tags cloned in pcDNA3; pcDNA3-GS2-NleG2-3 GS2 fused to EHEC O157:H7 NleG2-3 This study lacking N-terminal domain with C-terminal Flag and 6x-His tags cloned in pcDNA3; pcDNA3-GS2-NleG5-1 GS2 fused to EHEC O157:H7 NleG5-1 This study lacking N-terminal domain with C-terminal Flag and 6x-His tags cloned in pcDNA3; pcDNA3-GS2-NleL GS2 fused to EHEC O157:H7 NleL lacking β- This study helix domain with C-terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; pcDNA3-SidC-GS2 GS2 fused to This study terminal domain with N-terminal Flag and 6x- His tags cloned in pcDNA3; CMV promoter; pcDNA3-GS2-SlrP GS2 fused to Enterohemorrhagic This study domain with C-terminal Flag and 6x-His tags pcDNA3-GS2-SopA GS2 fused to This study helix domain with C-terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; pcDNA3-GS2-SspH1 GS2 fused to This study SspH1 lacking LRR domain with C-terminal Flag and 6x-His tags cloned in pcDNA3; pcDNA3-GS2-SspH2 GS2 fused to This study LRR domain with C-terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; pcDNA3-GS2-XopL GS2 fused to This study XopL lacking LRR domain with C-terminal Flag and 6x-His tags cloned in pcDNA3; pcDNA-βTrCP-GS2 GS2 fused to full length This study with C-terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; AmpR pcDNA3-GS2-CHIPΔTPR GS2 cloned in place of scFvR4 in pcDNA3- This study R4-CHIPΔTPR; CMV promoter; Kozak; pcDNA3-Slmb-GS2 GS2 cloned in place of vhhGFP4 in This study pcDNA3-NSlmb- vhhGFP4; CMV pcDNA3-GS2-SPOP GS2 fused to This study with C-terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; pcDNA3-VHL-GS2 GS2 fused to This study terminal Flag and 6x-His tags cloned in pcDNA3-GS5-IpaH9.8 GS5 fused to IpaH9.8 lacking LRR domain This study with C-terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; AmpR pcDNA3-GL4-IpaH9.8 GL4 fused to IpaH9.8 lacking LRR domain This study with C-terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; AmpR pcDNA3-GL6TpaH9.8 GL6 fused to IpaH9.8 lacking LRR domain This study with C-terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; AmpR pcDNA3-GL8-IpaH9.8 GL8 fused to IpaH9.8 lacking LRR domain This study with C-terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; AmpR pcDNA3-vhhGFP4-IpaH9.8 vhhGFP4 fused to IpaH9.8 lacking LRR This study domain with C- terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; pcDNA3-NSa5-IpaH9.8 NSa5 fused to IpaH9.8 lacking LRR domain This study with C-terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; AmpR pcDNA3-NSa5- NSa5 fused to IpaH9.8C337Alacking LRR This study IpaH9.8C337A domain with C- terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; pcDNA3-RasInll-IpaH9.8 RasInll fused to IpaH9.8 lacking LRR domain This study with C-terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; Kozak; AmpR pcDNA3-RasInll- RasInll fused to IpaH9.8C337Alacking LRR This study IpaH9.8C337A domain with C- terminal Flag and 6x-His tags cloned in pcDNA3; CMV promoter; mEmerald-C1 CMV promoter; KanR Laboratory stock mVenus-N1 CMV promoter; KanR Laboratory stock pcDNA3-mCerulean CMV promoter; Kozak; AmpR This study pcDNA3-CFP CMV promoter; Kozak; AmpR This study pcDNA3-sfGFP CMV promoter; Kozak; AmpR This study pcDH--mCherry CMV promoter; EF1α promoter; PuroR; AmpR Laboratory stock pPB-GS2-IpaH9.8 GS2-IpaH9.8 with C-terminal Flag and 6x-His This study tags cloned in pPB TetOn Hygro; CMV pPB-GS2-IpaH9.8C337A GS2-IpaH9.8C337Awith C-terminal Flag and 6x- This study His tags cloned in pPB TetOn Hygro; CMV promoter; SV40 promoter; AmpR pCDH1-ERK2-EGFP ERK2 fused to N-terminus of EGFP cloned in This study pCDH1-CMV- MCS-EF1α-Puro; CMV pCDHI-EGFP EGFP cloned in pCDH1-CMV-MCS- This study EF1α-Puro; CMV promoter; PurR, pCDH1-EGFP-HRasG12V HRasG12Vfused to C-terminus of EGFP This study cloned in pCDH1- CMV-MCS-EF1α-Puro; mEmerald-α-actinin-19 α-actinin fused to N-terminus of mEmerald; Laboratory stock CMV promoter; KanR pcDNA3.1(−)-α-synuclein- α-synuclein fused to the N-terminus of [10] EGFP EGFP cloned in pcDNA3.1; CMV mEmerald-FAK-5 FAK fused to C-terminus of mEmerald; CMV Laboratory stock pEGFP-C1 F-tractin-EGFP F-tractin fused to N-terminus of EGFP; CMV Addgene #58473 EGFR-mEmerald EGFR fused to N-terminus of mEmerald; Laboratory stock CMV promoter; AmpR pErbB2-EGFP ErbB2 fused to N-terminus of EGFP; CMV Addgene #39321 pcDNA3-ERK2-EGFP ERK2 fused to EGFP in pcDNA3; CMV This study promoter; Kozak; AmpR pLV pGK-H2B-EGFP H2B fused to C-terminus of EGFP; pGK Laboratory stock mEGFP-HRasG12V HRasG12V(constitutively active) fused to [11]; Addgene mEGFP cloned in pCI; CMV promoter; #18666 mEmerald-Muc1-FL Muc1 fused to N-terminus of mEmerald; CMV Laboratory stock pcDNA3-EGFP-NLS NLS sequence derived from C-terminus of This study SV40 fused to C- terminus of EGFP and cloned in pcDNA3; CMV promoter; AmpR mEmerald-Paxillin-22 Paxillin fused to N-terminus of mEmerald; Laboratory stock CMV promoter; KanR pcDNA3-SHP2-EGFP SHP2 fused to C-terminus of EGFP; CMV This study mEmerald-Vinculin-23 Vinculin fused to C-terminus of mEmerald; Laboratory stock CMV promoter; KanR pET28-EGFP EGFP with C-terminal 6x-His tag in This study pET28a(+); T7lac promoter; KanR pET28(+)-GS2 GS2 with C-terminal Flag and 6x-His tags This study in pET28a(+); T7lac promoter; KanR pET24d(+)-GS2-IpaH9.8 GS2 fused to IpaH9.8 lacking LRR domain This study with C-terminal Flag and 6x-His tags in pET24d(+)-IpaH9.8ALRR IpaH9.8 lacking LRR domain with C-terminal This study Flag and 6x-His tags in pET28a(+); T7lac pTriEX-3-GS2-IpaH9.8C337A GS2 fused to IpaH9.8 lacking LRR domain This study and containing Cys337Ala mutation in pTriEx-3; CMV promoter; T7 promoter; pGEM4Z/GS2-IpaH9.8/A64 In vitro transcription of GS2-IpaH9.8 with 3′ This study human globin UTR and 64 residue poly A pGEM4Z/GS2- In vitro transcription of GS2-IpaH9.8C337A This study IpaH9.8C337A/A64 with 3′ human globin UTR and 64 residue pGEM4Z/AS15- In vitro transcription of AS15-IpaH9.8 with 3′ This study IpaH9.8/A64 human globin UTR and 64 residue poly A [1] Caussinus et al., “Fluorescent Fusion Protein Knockout Mediated by Anti-GFP Nanobody,” [2] Yusa et al., “A Hyperactive PiggyBac Transposase for Mammalian Applications,” [3] Li et al., “Structurally Modulated Codelivery of siRNA and Argonautc 2 for Enhanced RNA interference,” [4] Boczkowski et al., “Induction of Tumor Immunity and Cytotoxic T Lymphocyte Responses Using Dendritic Cells Transfected With Messenger RNA Amplified From Tumor Cells.” [5] Koide et al., “Teaching an Old Scaffold New Tricks: Monobodies Constructed Using Alternative Surfaces of the FN3 Scaffold,” [6] Sha et al., “Dissection of the BCR-ABL Signaling Network Using Highly Specific Monobody Inhibitors to the SHP2 SH2 Domains,” [7] Cetin et al., “Raslns: Genetically Encoded Intrabodies of Activated Ras Proteins,” [8] Portnoff et al., “Ubiquibodies, Synthetic E3 Ubiquitin Ligases Endowed With Unnatural Substrate Specificity for Targeted Protein Silencing,” [9] Khokhlatchev et al., “Reconstitution of Mitogenactivated Protein Kinase Phosphorylation Cascades in Bacteria. Efficient Synthesis of Active Protein Kinases,” [10] Butler et al., “Bifunctional Anti-Nonamyloid Component α-synuclein Nanobodies are Protective In Situ” [11] Yasuda et al., “Supersensitive Ras Activation in Dendrites and Spines Revealed by Two-Photon Fluorescence Lifetime Imaging,” Example 1—Engineered IpaH9.8 Potently Silences GFP in Mammalian Cells
Bacterial E3 ubiquitin ligases evaluated in this study. E3 ubiquitin ligase Classification Organism Construction Ref.1 AvrPtoB U-box AvrPtoB1-436fused to N-terminus of GS2 [12] βTrCP F-box βTrCP1-568fused to N-terminus of GS2 [13] with (GlySer)10linker CHIP U-box CHIP128-303fused to C-terminus of GS2 [14] IpaH0722 NEL IpaH0722295-587fused to C-terminus of GS2 [15] IpaH1.4 NEL IpaH1.4285-575fused to C-terminus of GS2 [15] IpaH2.5 NEL IpaH2.5292-570fused to C-terminus of GS2 [15] IpaH4.5 NEL IpaH4.5296-574fused to C-terminus of GS2 [15] IpaH7.8 NEL IpaH7.8274-565fused to C-terminus of GS2 [15] IpaH9.8 NEL IpaH9.8254-545fused to C-terminus of GS2 [16] LegAU13 F-box LegAU131-50fused to N-terminus of GS2 [17] LegU1 F-box LegU11-56fused to N-terminus of GS2 [17] LubX U-box LubX1-215fused to N-terminus of GS2 [18] NleG2-3 U-box Enterohemorrhagic NleG2-390-191fused to C-terminus of GS2 [19] (EHEC) O157:H7 NleG5-1 U-box EHEC O157:H7 NleG5-1113-213fused to C-terminus of GS2 [19] NleL HECT EHEC O157:H7 NleL371-782fused to C-terminus of GS2 [20] SidC Unconventional SidC1-542fused to N-terminus of GS2 [21] SlrP NEL EHEC O157:H7 SlrP465-765fused to N-terminus of GS2 [22] SopA HECT SopA370-782fused to C-terminus of GS2 [23] SPOP F-box SPOP167-374fused to C-terminus of GS2 [24] SspH1 NEL SspH1404-701fused to N-terminus of GS2 [25] SspH2 NEL SspH2492-788fused to C-terminus of GS2 [26] VHL SCF-like ECV VHL1-213fused to N-terminus of GS2 [27] XopL Unconventional XopL474-660fused to C-terminus of GS2 [28]
1References listed in Table 2 provide clear proof or annotation of the catalytic domains of each E3 ubiquitin ligase.
Example 2—a Broad Range of Substrate Proteins is Degraded by GS2-IpaH9.8
Example 3—GS2-IpaH9.8-Mediated Proteome Editing is Flexible and Modular
Example 4—Delivery of mRNA Encoding GS2-IpaH9.8 Enables Proteome Editing in Mice
Example 5—Discussion of Examples 1-4