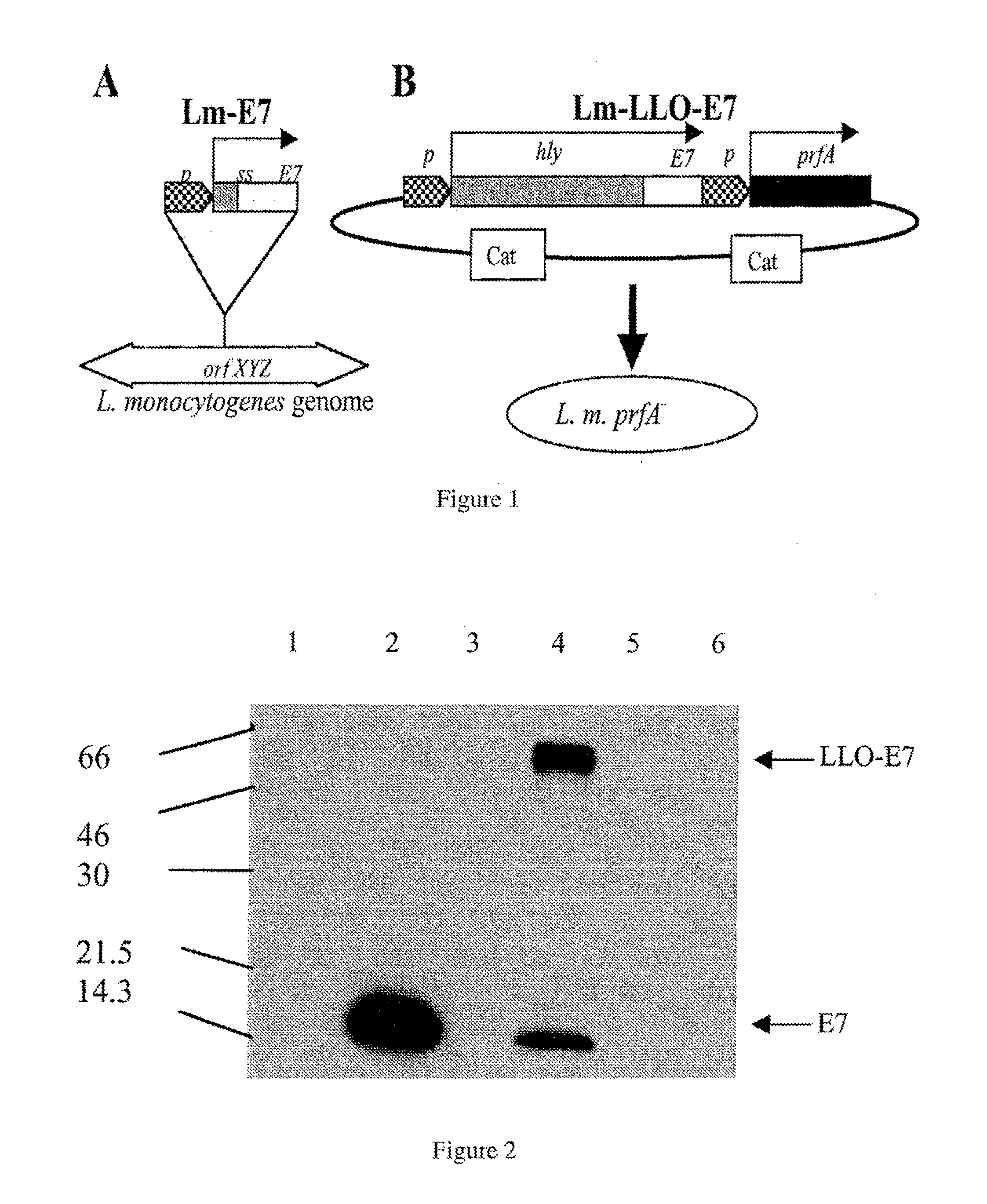
RECOMBINANT LISTERIA VACCINE STRAINS AND METHODS OF PRODUCING THE SAME
The present invention provides methods of treating, protecting against, and inducing an immune response against a tumor or cancer, comprising the step of administering to a subject a recombinant Persistent infection with high-oncogenic risk human papillomavirus (HR-HPV) types is recognized as a necessary, but not sufficient, cause of invasive carcinoma of the cervix (ICC) [1-3]. HPVs 16 and 18 are the most prevalent types in malignant lesions, accounting for over 70% of ICC and over 50% of high-grade precursor lesions. The HR-HPV E6 and E7 proteins are consistently expressed in dysplasias and carcinomas, disrupting the cell cycle regulatory proteins p53 and pRb, respectively. The obligatory expression of E6 and E7 by both dysplastic and invasive malignant lesions, as well as the viral origin of these proteins, make them excellent targets for HPV therapeutic vaccines. Lm has also a number of inherent advantages as a vaccine vector. The bacterium grows very efficiently in vitro without special requirements and it lacks LPS, which is a major toxicity factor in gram-negative bacteria, such as The PrfA protein controls the expression of a regulon comprising essential virulence genes required by Lm to colonize its vertebrate hosts; hence the prfA mutation strongly impairs PrfA ability to activate expression of PrfA-dependent virulence genes. The present invention addresses this concern by providing a prfA mutant In one embodiment, the present invention relates to a recombinant In one embodiment, the present invention relates to a recombinant In one embodiment, the present invention relates to a method for inducing an immune response against a tumor or a cancer in a subject, the method comprising the step of administering to said subject a recombinant Other features and advantages of the present invention will become apparent from the following detailed description examples and figures. It should be understood, however, that the detailed description and the specific examples while indicating preferred embodiments of the invention are given by way of illustration only, since various changes and modifications within the spirit and scope of the invention will become apparent to those skilled in the art from this detailed description. The subject matter regarded as the invention is particularly pointed out and distinctly claimed in the concluding portion of the specification. The invention, however, both as to organization and method of operation, together with objects, features, and advantages thereof, may best be understood by reference to the following detailed description when read with the accompanying drawings in which: It will be appreciated that for simplicity and clarity of illustration, elements shown in the figures have not necessarily been drawn to scale. For example, the dimensions of some of the elements may be exaggerated relative to other elements for clarity. Further, where considered appropriate, reference numerals may be repeated among the figures to indicate corresponding or analogous elements. The present invention provides, in one embodiment, a recombinant The present invention further provides immunogenic compositions comprising a recombinant The present invention also provides methods for inducing an anti-disease cytotoxic T-cell (CTL) response in a subject and treating disorders, and symptoms associated with said disease comprising administering a recombinant In another embodiment, a recombinant It will be well appreciated by a skilled artisan that the term “Attenuated gene” may encompass a gene that mediates toxicity, pathology, or virulence, to a host, growth within the host, or survival within the host, where the gene is mutated in a way that mitigates, reduces, or eliminates the toxicity, pathology, or virulence. The reduction or elimination can be assessed by comparing the virulence or toxicity mediated by the mutated gene with that mediated by the non-mutated (or parent) gene. “Mutated gene” encompasses deletions, point mutations, and frameshift mutations in regulatory regions of the gene, coding regions of the gene, non-coding regions of the gene, or any combination thereof. In one embodiment, provided herein is a method for inducing an immune response against a tumor or a cancer in a subject, the method comprising the step of administering to said subject a composition comprising a recombinant In one embodiment, the present invention provides a method of treating a tumor or cancer in a subject, comprising the step of administering to the subject a composition comprising a recombinant In one embodiment, the methods provided herein further comprise the step of boosting a subject with a composition comprising a recombinant In one embodiment, the fragment thereof in the context of LLO proteins and ActA proteins provided herein refer to a peptide or polypeptide comprising an amino acid sequence of at least 5 contiguous amino acid residues of the LLO or ActA proteins. In another embodiment, the term refers to a peptide or polypeptide comprising an amino acid sequence of at least of at least 10 contiguous amino acid residues, at least 15 contiguous amino acid residues, at least 20 contiguous amino acid residues, at least 25 contiguous amino acid residues, at least 40 contiguous amino acid residues, at least 50 contiguous amino acid residues, at least 60 contiguous amino residues, at least 70 contiguous amino acid residues, at least 80 contiguous amino acid residues, at least 90 contiguous amino acid residues, at least 100 contiguous amino acid residues, at least 125 contiguous amino acid residues, at least 150 contiguous amino acid residues, at least 175 contiguous amino acid residues, at least 200 contiguous amino acid residues, at least 250 contiguous amino acid residues of the amino acid sequence, at least 300 contiguous amino acid residues, at least 350 contiguous amino acid residues of, at least 400 contiguous amino acid residues, or at least 450 contiguous amino acid residues of an LLO or ActA protein or polypeptide. In another embodiment, a “fragment” is a functional fragment that comprises a biological activity (e.g. to elicit an immune response against a heterologous antigen expressed by a tumor cell, either when administered alone or when administered in the context of a fusion protein as further described herein. In another embodiment, the fragment is functional in a non-fused form. The present invention, in certain embodiments, provides codon optimization of a nucleic acid heterologous to An N-terminal LLO protein fragment and heterologous antigen provided herein are, in one embodiment, fused directly to one another. In another embodiment, the genes encoding the N-terminal LLO protein fragment and the heterologous antigen are fused directly to one another. In another embodiment, the N-terminal LLO protein fragment and the heterologous antigen are attached via a linker peptide. In another embodiment, the N-terminal LLO protein fragment and the heterologous antigen are attached via a heterologous peptide. In another embodiment, the N-terminal LLO protein fragment is N-terminal to the heterologous antigen. In another embodiment, the N-terminal LLO protein fragment is the N-terminal-most portion of the fusion protein. Each possibility represents a separate embodiment of the present invention. As provided herein, recombinant In another embodiment, the present invention provides a method of treating a tumor or cancer in a subject, comprising the step of administering to the subject a recombinant In one embodiment, the terms “recombinant polypeptide” and “fusion protein” are used interchangeably herein. In another embodiment, the present invention provides a method of protecting a subject against a tumor or cancer, comprising the step of administering to the subject a recombinant In another embodiment, the present invention provides a method for inducing an immune response against a tumor or cancer in a subject, comprising the step of administering to the subject a recombinant In another embodiment, the present invention provides a method of treating a tumor or cancer in a subject, comprising the step of administering to the subject a recombinant In another embodiment, the present invention provides a method of protecting a subject against a tumor or cancer, comprising the step of administering to the subject a recombinant In another embodiment, the present invention provides a method for inducing an immune response against a tumor or cancer in a subject, comprising the step of administering to the subject a recombinant The N-terminal ActA protein fragment and the heterologous antigen are, in another embodiment, fused directly to one another. In another embodiment, the genes encoding the N-terminal ActA protein fragment and heterologous antigen are fused directly to one another. In another embodiment, the N-terminal ActA protein fragment and heterologous antigen are attached via a linker peptide. In another embodiment, the N-terminal ActA protein fragment and heterologous antigen are attached via a heterologous peptide. In another embodiment, the N-terminal ActA protein fragment is N-terminal to the heterologous antigen. In another embodiment, the N-terminal ActA protein fragment is the N-terminal-most portion of the fusion protein. Each possibility represents a separate embodiment of the present invention. In another embodiment, the present invention provides a method of inducing an immune response against a tumor or cancer in a subject, comprising the step of administering to the subject a recombinant The PEST amino acid sequence-containing peptide and heterologous antigen are, in another embodiment, fused directly to one another. In another embodiment, the genes encoding the PEST amino acid sequence-containing peptide and heterologous antigen are fused directly to one another. In another embodiment, the PEST amino acid sequence-containing peptide and heterologous antigen are attached via a linker peptide. In another embodiment, the PEST amino acid sequence-containing peptide and heterologous antigen are attached via a heterologous peptide. In another embodiment, the PEST amino acid sequence-containing peptide is N-terminal to the heterologous antigen. In another embodiment, the PEST amino acid sequence-containing peptide is the N-terminal-most portion of the fusion protein. Each possibility represents a separate embodiment of the present invention. In another embodiment, the present invention provides a method for vaccinating a subject against an HPV, comprising the step of administering to the subject a prfA mutant recombinant In one embodiment, provided herein is a method of increasing a ratio of T effector cells to regulatory T cells (Tregs) in the spleen and tumor microenvironments of a subject, comprising administering the immunogenic composition provided herein. In another embodiment, increasing a ratio of T effector cells to regulatory T cells (Tregs) in the spleen and tumor microenvironments in a subject allows for a more profound anti-tumor response in the subject. In one embodiment, a mutant PrfA protein provided herein comprises a D133V amino acid mutation. In another embodiment, the mutant PrfA protein consists of a D133V amino acid mutation. In another embodiment, a nucleic acid comprising an open reading frame encoding a mutant PrfA protein provided herein is in a plasmid in said recombinant In one embodiment, a prfA mutant recombinant In one embodiment, a mutant PrfA protein provided herein complements a genomic deletion, inactivation or mutation in the prfA gene in a recombinant In one embodiment, a wild-type PrfA protein is encoded by the following wild-type nucleic acid sequence set forth in SEQ ID NO: 31. In one embodiment, a wild-type PrfA protein comprises an amino acid sequence set forth in SEQ ID NO: 32. In one embodiment, a nucleic acid sequence encoding a mutant prfA sequence is set forth in SEQ ID NO: 33. In one embodiment, a mutant PrfA protein provided herein comprises an amino acid sequence set forth in SEQ ID NO: 34. M N A Q A E E F K K Y L E T N G I K P K Q F H K K E L I F N Q W D P Q E Y C I F L Y D G I T K L T S I S E N G T I M N L Q Y Y K G A F V I M S G F I D T E T S V G Y Y N L E V I S E Q A T A Y V I K I N E L K E L L S K N L T H F F Y V F Q T L Q K Q V S Y S L A K F N V F S I N G K L G S I C G Q L L I L T Y V Y G K E T P D G I K I T L D N L T M Q E L G Y S S G I A H S S A V S R I I S K L K Q E K V I V Y K N S C F Y V Q N R D Y L K R Y A P K L D E W F Y L A C P A T W G K L N (SEQ ID NO: 34). In another embodiment, SEQ ID NO: 34 represents a mutant PrfA protein comprising a D133V mutation. In another embodiment, a mutant PrfA protein is homologous to SEQ ID NO: 34 and comprises a D133V mutation. In another embodiment, a mutant PrfA protein is at least 90% homologous with SEQ ID NO: 34 and comprises a D133V mutation. In another embodiment, a mutant PrfA protein is at least 85% homologous with SEQ ID NO: 34, and comprises a D133V mutation. In another embodiment, the subject is at risk for developing an HPV-mediated carcinogenesis (e.g. a cervical, head and neck or anal cancer). In another embodiment, the subject is HPV-positive. In another embodiment, the subject exhibits cervical intraepithelial neoplasia. In another embodiment, the subject exhibits a squamous intraepithelial lesion. In another embodiment, the subject exhibits a dysplasia in the cervix. The HPV that is the target of methods of the present invention is, in another embodiment, an HPV 16. In another embodiment, the HPV is an HPV-18. In another embodiment, the HPV is selected from HPV-16 and HPV-18. In another embodiment, the HPV is an HPV-31. In another embodiment, the HPV is an HPV-35. In another embodiment, the HPV is an HPV-39. In another embodiment, the HPV is an HPV-45. In another embodiment, the HPV is an HPV-51. In another embodiment, the HPV is an HPV-52. In another embodiment, the HPV is an HPV-58. In another embodiment, the HPV is a high-risk HPV type. In another embodiment, the HPV is a mucosal HPV type. Each possibility represents a separate embodiment of the present invention. In another embodiment, the present invention provides a method of vaccinating a subject against an antigen of interest, the method comprising the step of intravenously administering to the subject an immunogenic composition, comprising a fusion of an immunogenic peptide to the antigen of interest, wherein the immunogenic peptide is selected from (a) an N-terminal fragment of an LLO protein; (b) an ActA protein or N-terminal fragment thereof; and (c) a PEST amino acid sequence-containing peptide, thereby vaccinating a subject against an antigen of interest. In another embodiment, the present invention provides a method of vaccinating a subject against an antigen of interest, the method comprising the step of administering intravenously to the subject a recombinant In another embodiment, the present invention provides a method of inducing a CTL response in a subject against an antigen of interest, the method comprising the step of administering to the subject a recombinant As provided herein, recombinant In another embodiment, the present invention provides a method for inducing a regression of a cancer in a subject, comprising the step of administering to the subject a composition comprising a recombinant In another embodiment, the present invention provides a method for reducing an incidence of relapse of a cancer in a subject, comprising the step of administering to the subject a composition comprising a recombinant In another embodiment, the present invention provides a method for suppressing a formation of a tumor in a subject, comprising the step of administering to the subject a composition comprising recombinant In another embodiment, the present invention provides a method for inducing a remission of a cancer in a subject, comprising the step of administering to the subject a composition comprising a recombinant In another embodiment, the present invention provides a method for impeding a growth of a tumor in a subject, comprising the step of administering to the subject a composition comprising a recombinant In another embodiment, the present invention provides a method for reducing a size of a tumor in a subject, comprising the step of administering to the subject a composition comprising a recombinant In one embodiment, a disease is an infectious disease, an autoimmune disease, a respiratory disease, a pre-cancerous condition or a cancer. It will be well appreciated by the skilled artisan that the term “pre-cancerous condition” may encompass dysplasias, preneoplastic nodules; macroregenerative nodules (MRN); low-grade dysplastic nodules (LG-DN); high-grade dysplastic nodules (HG-DN); biliary epithelial dysplasia; foci of altered hepatocytes (FAH); nodules of altered hepatocytes (NAH); chromosomal imbalances; aberrant activation of telomerase; re-expression of the catalytic subunit of telomerase; expression of endothelial cell markers such as CD31, CD34, and BNH9 (see, e.g., Terracciano and Tomillo (2003) Pathologica 95:71-82; Su and Bannasch (2003) Toxicol. Pathol. 31:126-133; Rocken and Carl-McGrath (2001) Dig. Dis. 19:269-278; Kotoula, et al. (2002) Liver 22:57-69; Frachon, et al. (2001) J. Hepatol. 34:850-857; Shimonishi, et al. (2000) J. Hepatobiliary Pancreat. Surg. 7:542-550; Nakanuma, et al. (2003) J. Hepatobiliary Pancreat. Surg. 10:265-281). Methods for diagnosing cancer and dysplasia are disclosed (see, e.g., Riegler (1996) Semin Gastrointest. Dis. 7:74-87; Benvegnu, et al. (1992) Liver 12:80-83; Giannini, et al. (1987) Hepatogastroenterol. 34:95-97; Anthony (1976) Cancer Res. 36:2579-2583). In one embodiment, an infectious disease is one caused by, but not limited to, any one of the following pathogens: BCG/Tuberculosis, Malaria, In another embodiment, the infectious disease is a livestock infectious disease. In another embodiment, livestock diseases can be transmitted to man and are called “zoonotic diseases.” In another embodiment, these diseases include, but are not limited to, Foot and mouth disease, West Nile Virus, rabies, canine parvovirus, feline leukemia virus, equine influenza virus, infectious bovine rhinotracheitis (IBR), pseudorabies, classical swine fever (CSF), IBR, caused by bovine herpesvirus type 1 (BHV-1) infection of cattle, and pseudorabies (Aujeszky's disease) in pigs, toxoplasmosis, anthrax, vesicular stomatitis virus, In another embodiment, the disease provided herein is a respiratory or inflammatory disease. In another embodiment, the respiratory or inflammatory disease is chronic obstructive pulmonary disease (COPD). In another embodiment, the disease is asthma. In one embodiment, live attenuated In one embodiment, a disease is a cancer or a tumor. In one embodiment, the tumor is cancerous. In another embodiment, the cancer is breast cancer. In another embodiment, the cancer is a cervical cancer. In another embodiment, the cancer is a Her2 containing cancer. In another embodiment, the cancer is a melanoma. In another embodiment, the cancer is pancreatic cancer. In another embodiment, the cancer is ovarian cancer. In another embodiment, the cancer is gastric cancer. In another embodiment, the cancer is a carcinomatous lesion of the pancreas. In another embodiment, the cancer is pulmonary adenocarcinoma. In another embodiment, it is a glioblastoma multiforme. In another embodiment, the cancer is colorectal adenocarcinoma. In another embodiment, the cancer is pulmonary squamous adenocarcinoma. In another embodiment, the cancer is gastric adenocarcinoma. In another embodiment, the cancer is an ovarian surface epithelial neoplasm (e.g. a benign, proliferative or malignant variety thereof). In another embodiment, the cancer is an oral squamous cell carcinoma. In another embodiment, the cancer is non-small-cell lung carcinoma. In another embodiment, the cancer is an endometrial carcinoma. In another embodiment, the cancer is a bladder cancer. In another embodiment, the cancer is a head and neck cancer. In another embodiment, the cancer is a prostate carcinoma. In another embodiment, the cancer is oropharyngeal cancer. In another embodiment, the cancer is lung cancer. In another embodiment, the cancer is anal cancer. In another embodiment, the cancer is colorectal cancer. In another embodiment, the cancer is esophageal cancer. The cervical tumor targeted by methods of the present invention is, in another embodiment, a squamous cell carcinoma. In another embodiment, the cervical tumor is an adenocarcinoma. In another embodiment, the cervical tumor is an adenosquamous carcinoma. In another embodiment, the cervical tumor is a small cell carcinoma. In another embodiment, the cervical tumor is any other type of cervical tumor known in the art. A cervical tumor targeted by methods of the present invention is, in one embodiment, a squamous cell carcinoma. In another embodiment, the cervical tumor is an adenocarcinoma. In another embodiment, the cervical tumor is an adenosquamous carcinoma. In another embodiment, the cervical tumor is a small cell carcinoma. In another embodiment, the cervical tumor is any other type of cervical tumor known in the art. Each possibility represents a separate embodiment of the present invention. In one embodiment, the terms “tumor antigen” “antigenic polypeptide,” or “foreign antigen” are used interchangeably herein and include tumor antigens, tumor-associated antigens, angiogenic antigens, or infectious disease antigens. In another embodiment, an antigen provided herein is a self-antigen that is present in the host but the host does not elicit an immune response against it because of immunologic tolerance. In one embodiment, the antigen is Human Papilloma Virus-E7 (HPV-E7) antigen, which in one embodiment, is from HPV16 (in one embodiment, GenBank Accession No. AAD33253) and in another embodiment, from HPV18 (in one embodiment, GenBank Accession No. P06788). In another embodiment, the antigenic polypeptide is HPV-E6, which in one embodiment, is from HPV16 (in one embodiment, GenBank Accession No. AAD33252, AAM51854, AAM51853, or AAB67615) and in another embodiment, from HPV18 (in one embodiment, GenBank Accession No. P06463). In another embodiment, the antigenic polypeptide is a Her/2-neu antigen. In another embodiment, the antigenic polypeptide is Prostate Specific Antigen (PSA) (in one embodiment, GenBank Accession No. CAD30844, CAD54617, AAA58802, or NP_001639). In another embodiment, the antigenic polypeptide is Stratum Corneum Chymotryptic Enzyme (SCCE) antigen (in one embodiment, GenBank Accession No. AAK69652, AAK69624, AAG33360, AAF01139, or AAC37551). In another embodiment, the antigenic polypeptide is Wilms tumor antigen 1, which in another embodiment is WT-1 Telomerase (GenBank Accession. No. P49952, P22561, NP_659032, CAC39220.2, or EAW68222.1). In another embodiment, the antigenic polypeptide is hTERT or Telomerase (GenBank Accession. No. NM003219 (variant 1), NM198255 (variant 2), NM 198253 (variant 3), or NM 198254 (variant 4). In another embodiment, the antigenic polypeptide is Proteinase 3 (in one embodiment, GenBank Accession No. M29142, M75154, M96839, X55668, NM 00277, M96628 or X56606). In another embodiment, the antigenic polypeptide is Tyrosinase Related Protein 2 (TRP2) (in one embodiment, GenBank Accession No. NP_001913, ABI73976, AAP33051, or Q95119). In another embodiment, the antigenic polypeptide is High Molecular Weight Melanoma Associated Antigen (HMW-MAA) (in one embodiment, GenBank Accession No. NP_001888, AAI28111, or AAQ62842). In another embodiment, the antigenic polypeptide is Testisin (in one embodiment, GenBank Accession No. AAF79020, AAF79019, AAG02255, AAK29360, AAD41588, or NP_659206). In another embodiment, the antigenic polypeptide is NY-ESO-1 antigen (in one embodiment, GenBank Accession No. CAA05908, P78358, AAB49693, or NP_640343). In another embodiment, the antigenic polypeptide is PSCA (in one embodiment, GenBank Accession No. AAH65183, NP_005663, NP_082492, 043653, or CAB97347). In another embodiment, the antigenic polypeptide is Interleukin (IL) 13 Receptor alpha (in one embodiment, GenBank Accession No. NP_000631, NP_001551, NP_032382, NP_598751, NP_001003075, or NP_999506). In another embodiment, the antigenic polypeptide is Carbonic anhydrase IX (CAIX) (in one embodiment, GenBank Accession No. CAI13455, CAI10985, EAW58359, NP_001207, NP_647466, or NP_001101426). In another embodiment, the antigenic polypeptide is carcinoembryonic antigen (CEA) (in one embodiment, GenBank Accession No. AAA66186, CAA79884, CAA66955, AAA51966, AAD15250, or AAA51970.). In another embodiment, the antigenic polypeptide is MAGE-A (in one embodiment, GenBank Accession No. NP_786885, NP_786884, NP_005352, NP_004979, NP_005358, or NP—005353). In another embodiment, the antigenic polypeptide is survivin (in one embodiment, GenBank Accession No. AAC51660, AAY15202, ABF60110, NP_001003019, or NP_001082350). In another embodiment, the antigenic polypeptide is GP100 (in one embodiment, GenBank Accession No. AAC60634, YP_655861, or AAB31176). In another embodiment, the antigenic polypeptide is any other antigenic polypeptide known in the art. In another embodiment, the antigenic peptide of the compositions and methods of the present invention comprise an immunogenic portion of the antigenic polypeptide. Each possibility represents a separate embodiment of the present invention. In another embodiment, the antigen is telomerase (TERT). In another embodiment, the antigen is LMP-1. In another embodiment, the antigen is p53. In another embodiment, the antigen is mesothelin. In another embodiment, the antigen is EGFRVIII. In another embodiment, the antigen is carboxic anhydrase IX (CAIX). In another embodiment, the antigen is PSMA. In another embodiment, the antigen is HMW-MAA. In another embodiment, the antigen is HIV-1 Gag. In another embodiment, the antigen is Tyrosinase related protein 2. In another embodiment, the antigen is selected from Her-2, HIV-1 Gag, LMP-1, p53, PSMA, carcinoembryonic antigen (CEA), LMP-1,kallikrein-related peptidase 3 (KLK3), KLK9, Muc, Tyrosinase related protein 2, Muc1, FAP, IL-13R alpha 2, PSA (prostate-specific antigen), gp-100, heat-shock protein 70 (HSP-70), beta-HCG, EGFR-III, Granulocyte colony-stimulating factor (G-CSF), Angiogenin, Angiopoietin-1, Del-1, Fibroblast growth factors: acidic (aFGF) or basic (bFGF), Follistatin, Granulocyte colony-stimulating factor (G-CSF), Hepatocyte growth factor (HGF)/scatter factor (SF), Interleukin-8 (IL-8), Leptin, Midkine, Placental growth factor, Platelet-derived endothelial cell growth factor (PD-ECGF), Platelet-derived growth factor-BB (PDGF-BB), Pleiotrophin (PTN), Progranulin, Proliferin, Transforming growth factor-alpha (TGF-alpha), Transforming growth factor-beta (TGF-beta), Tumor necrosis factor-alpha (TNF-alpha), Vascular endothelial growth factor (VEGF)/vascular permeability factor (VPF), VEGFR, VEGFR2 (KDR/FLK-1) or a fragment thereof, FLK-1 or an epitope thereof, FLK-E1, FLK-E2, FLK-I1, endoglin or a fragment thereof, Neuropilin 1 (NRP-1), Angiopoietin 1 (Ang1), Tie2, Platelet-derived growth factor (PDGF), Platelet-derived growth factor receptor (PDGFR), Transforming growth factor-beta (TGF-β), endoglin, TGF-β receptors, monocyte chemotactic protein-1 (MCP-1), VE-cadherin, CD31, ephrin, ICAM-1, V-CAM-1, VAP-1, E-selectin, plasminogen activators, plasminogen activator inhibitor-1, Nitric oxide synthase (NOS), COX-2, AC133, or Id1/Id3, Angiopoietin 3, Angiopoietin 4, Angiopoietin 6, CD105, EDG, HHT1, ORW, ORW1 or a TGFbeta co-receptor, or a combination thereof. In another embodiment, the antigen is a chimeric Her2/neu antigen as disclosed in US Patent Application Publication No. 2011/0142791, which is incorporated by reference herein in its entirety. The use of fragments of antigens provided herein is also encompassed by the present invention. In another embodiment, the tumor antigen provided herein is a tumor-associated antigen, which in one embodiment, is one of the following tumor antigens: a MAGE (Melanoma-Associated Antigen E) protein, e.g. MAGE 1, MAGE 2, MAGE 3, MAGE 4, a tyrosinase; a mutant ras protein; a mutant p53 protein; p97 melanoma antigen, a ras peptide or p53 peptide associated with advanced cancers; the HPV 16/18 antigens associated with cervical cancers, KLH antigen associated with breast carcinoma, CEA (carcinoembryonic antigen) associated with colorectal cancer, a MART1 antigen associated with melanoma, or the PSA antigen associated with prostate cancer. In another embodiment, the antigen for the compositions and methods provided herein are melanoma-associated antigens, which in one embodiment are TRP-2, MAGE-1, MAGE-3, gp-100, tyrosinase, HSP-70, beta-HCG, or a combination thereof. It is to be understood that a skilled artisan would be able to use any heterologous antigen not mentioned herein but known in the art for use in the methods and compositions provided herein. It is also to be understood that the present invention provides, but is not limited by, an attenuated In one embodiment, vascular endothelial growth factor (VEGF) is an important signaling protein involved in both vasculogenesis (the formation of the embryonic circulatory system) and angiogenesis (the growth of blood vessels from pre-existing vasculature). In one embodiment, VEGF activity is restricted mainly to cells of the vascular endothelium, although it does have effects on a limited number of other cell types (e.g. stimulation monocyte/macrophage migration). In vitro, VEGF has been shown to stimulate endothelial cell mitogenesis and cell migration. VEGF also enhances microvascular permeability and is sometimes referred to as vascular permeability factor. In one embodiment, all of the members of the VEGF family stimulate cellular responses by binding to tyrosine kinase receptors (the VEGFRs) on the cell surface, causing them to dimerize and become activated through transphosphorylation. The VEGF receptors have an extracellular portion consisting of 7 immunoglobulin-like domains, a single transmembrane spanning region and an intracellular portion containing a split tyrosine-kinase domain. In one embodiment, VEGF-A is a VEGFR-2 (KDR/Flk-1) ligand as well as a VEGFR-1 (Flt-1) ligand. In one embodiment, VEGFR-mediates almost all of the known cellular responses to VEGF. The function of VEGFR-1 is less well defined, although it is thought to modulate VEGFR-2 signaling, in one embodiment, via sequestration of VEGF from VEGFR-2 binding, which in one embodiment, is particularly important during vasculogenesis in the embryo. In one embodiment, VEGF-C and VEGF-D are ligands of the VEGFR-3 receptor, which in one embodiment, mediates lymphangiogenesis. In one embodiment, the antigen of the present invention is a VEGF receptor or a fragment thereof, which in one embodiment, is a VEGFR-2 and, in another embodiment, a VEGFR-1, and, in another embodiment, VEGFR-3. In one embodiment, vascular Endothelial Growth Factor Receptor 2 (VEGFR2) is highly expressed on activated endothelial cells (ECs) and participates in the formation of new blood vessels. In one embodiment, VEGFR2 binds all 5 isoforms of VEGF. In one embodiment, signaling of VEGF through VEGFR2 on ECs induces proliferation, migration, and eventual differentiation. In one embodiment, the mouse homologue of VEGFR2 is the fetal liver kinase gene-1 (Flk-1), which is a strong therapeutic target, and has important roles in tumor growth, invasion, and metastasis. In one embodiment, VEGFR2 is also referred to as kinase insert domain receptor (a type III receptor tyrosine kinase) (KDR), cluster of differentiation 309 (CD309), FLK1, Ly73, Krd-1, VEGFR, VEGFR-2, or 6130401C07. In other embodiments, the antigen is derived from a fungal pathogen, bacteria, parasite, helminth, or viruses. In other embodiments, the antigen is selected from tetanus toxoid, hemagglutinin molecules from influenza virus, diphtheria toxoid, HIV gp120, HIV gag protein, IgA protease, insulin peptide B, In other embodiments, the antigen is associated with one of the following diseases; cholera, diphtheria, In another embodiment, an HPV E6 antigen is utilized instead of or in addition to an E7 antigen in a method of the present invention for treating, protecting against, or inducing an immune response against a cervical cancer. In another embodiment, an ActA protein fragment is utilized instead of or in addition to an LLO fragment in a method of the present invention for treating, protecting against, or inducing an immune response against a cervical cancer. In another embodiment, a PEST amino acid sequence-containing protein fragment is utilized instead of or in addition to an LLO fragment in a method of the present invention for treating, protecting against, or inducing an immune response against a cervical cancer. In another embodiment, the present invention provides a method for inducing an anti-E7 cytotoxic T cell (CTL) response in a subject, comprising the step of administering to the subject a recombinant In another embodiment, the present invention provides a method of treating or ameliorating an HPV-mediated disease, disorder, or symptom in a subject, comprising the step of administering to the subject a recombinant In another embodiment, the HPV-mediated disease, disorder, or symptom is genital warts. In another embodiment, the HPV-mediated disease, disorder, or symptom is non-genital warts. In another embodiment, the HPV-mediated disease, disorder, or symptom is a respiratory papilloma. In another embodiment, the HPV-mediated disease, disorder, or symptom is any other HPV-mediated disease, disorder, or symptom known in the art. Each possibility represents a separate embodiment of the present invention. The antigen of methods and compositions of the present invention is, in another embodiment, an HPV E7 protein. In another embodiment, the antigen is an HPV E6 protein. In another embodiment, the antigen is any other HPV protein known in the art. Each possibility represents a separate embodiment of the present invention. “E7 antigen” refers, in another embodiment, to an E7 protein. In another embodiment, the term refers to an E7 fragment. In another embodiment, the term refers to an E7 peptide. In another embodiment, the term refers to any other type of E7 antigen known in the art. Each possibility represents a separate embodiment of the present invention. The E7 protein of methods and compositions of the present invention is, in another embodiment, an HPV 16 E7 protein. In another embodiment, the E7 protein is an HPV-18 E7 protein. In another embodiment, the E7 protein is an HPV-31 E7 protein. In another embodiment, the E7 protein is an HPV-35 E7 protein. In another embodiment, the E7 protein is an HPV-39 E7 protein. In another embodiment, the E7 protein is an HPV-45 E7 protein. In another embodiment, the E7 protein is an HPV-51 E7 protein. In another embodiment, the E7 protein is an HPV-52 E7 protein. In another embodiment, the E7 protein is an HPV-58 E7 protein. In another embodiment, the E7 protein is an E7 protein of a high-risk HPV type. In another embodiment, the E7 protein is an E7 protein of a mucosal HPV type. Each possibility represents a separate embodiment of the present invention. “E6 antigen” refers, in another embodiment, to an E6 protein. In another embodiment, the term refers to an E6 fragment. In another embodiment, the term refers to an E6 peptide. In another embodiment, the term refers to any other type of E6 antigen known in the art. Each possibility represents a separate embodiment of the present invention. The E6 protein of methods and compositions of the present invention is, in another embodiment, an HPV 16 E6 protein. In another embodiment, the E6 protein is an HPV-18 E6 protein. In another embodiment, the E6 protein is an HPV-31 E6 protein. In another embodiment, the E6 protein is an HPV-35 E6 protein. In another embodiment, the E6 protein is an HPV-39 E6 protein. In another embodiment, the E6 protein is an HPV-45 E6 protein. In another embodiment, the E6 protein is an HPV-51 E6 protein. In another embodiment, the E6 protein is an HPV-52 E6 protein. In another embodiment, the E6 protein is an HPV-58 E6 protein. In another embodiment, the E6 protein is an E6 protein of a high-risk HPV type. In another embodiment, the E6 protein is an E6 protein of a mucosal HPV type. Each possibility represents a separate embodiment of the present invention. In one embodiment, combinations of the E6 and E7 antigens are contemplated to fall within the scope of a “heterologous antigen” provided herein. The immune response induced by methods and compositions of the present invention is, in another embodiment, a T cell response. In another embodiment, the immune response comprises a cytotoxic T cell response. In another embodiment, the immune response comprises a T cell response. In another embodiment, the response is a CD8+ T cell response. In another embodiment, the response comprises a CD8+ T cell response. Each possibility represents a separate embodiment of the present invention. The N-terminal LLO protein fragment of methods and compositions of the present invention comprises, in another embodiment, SEQ ID No: 2. In another embodiment, the fragment comprises an LLO signal peptide. In another embodiment, the fragment comprises SEQ ID No: 2. In another embodiment, the fragment consists approximately of SEQ ID No: 2. In another embodiment, the fragment consists essentially of SEQ ID No: 2. In another embodiment, the fragment corresponds to SEQ ID No: 2. In another embodiment, the fragment is homologous to SEQ ID No: 2. In another embodiment, the fragment is homologous to a fragment of SEQ ID No: 2. The ΔLLO used in some of the Examples was 416 AA long (exclusive of the signal sequence), as 88 residues from the amino terminus which is inclusive of the activation domain containing cysteine 484 were truncated. It will be clear to those skilled in the art that any ΔLLO without the activation domain, and in particular without cysteine 484, are suitable for methods and compositions of the present invention. In another embodiment, fusion of an E7 and/or E6 antigen to any ΔLLO, including a PEST amino acid AA sequence, SEQ ID NO: 1, enhances cell mediated and anti-tumor immunity of the antigen. Each possibility represents a separate embodiment of the present invention. The LLO protein utilized to construct vaccines of the present invention has, in another embodiment, the sequence: MKKIMLVFITLILVSLPIAQQTEAKDASAFNKENSISSMAPPASPPASPKTPIEKKHADE IDKYIQGLDYNKNNVLVYHGDAVTNVPPRKGYKDGNEYIVVEKKKKSINQNNADIQ VVNAISSLTYPGALVKANSELVENQPDVLPVKRDSLTLSIDLPGMTNQDNKIVVKNA TKSNVNNAVNTLVERWNEKYAQAYPNVSAKIDYDDEMAYSESQLIAKFGTAFKAV NNSLNVNFGAISEGKMQEEVISFKQIYYNVNVNEPTRPSRFFGKAVTKEQLQALGVN AENPPAYISSVAYGRQVYLKLSTNSHSTKVKAAFDAAVSGKSVSGDVELTNIIKNSSF KAVIYGGSAKDEVQIIDGNLGDLRDILKKGATFNRETPGVPIAYTTNFLKDNELAVIK NNSEYIETTSKAYTDGKINIDHSGGYVAQFNISWDEVNYDPEGNEIVQHKNWSENNK SKLAHFTSSIYLPGNARNINVYAKECTGLAWEWWRTVIDDRNLPLVKNRNISIWGTT LYPKYSNKVDNPIE (GenBank Accession No. P13128; SEQ ID NO: 3; nucleic acid sequence is set forth in GenBank Accession No. X15127). The first 25 AA of the proprotein corresponding to this sequence are the signal sequence and are cleaved from LLO when it is secreted by the bacterium. Thus, in this embodiment, the full length active LLO protein is 504 residues long. In another embodiment, a full length LLO protein has an amino acid sequence of any full length wild-type LLO protein known in the art. In another embodiment, SEQ ID NO: 3 is used as the source of the LLO fragment incorporated in a vaccine of the present invention. Each possibility represents a separate embodiment of the present invention. In another embodiment, the N-terminal fragment of an LLO protein utilized in compositions and methods of the present invention has the sequence: In another embodiment, the LLO fragment has the sequence: In one embodiment, “Listeriolysin O protein,” or “LLO protein,” refer to a wild-type LLO protein unless stated to be a fragment of the same. In another embodiment, “truncated LLO” or “ΔLLO” refers to a fragment of LLO that comprises the PEST amino acid domain. In another embodiment, the terms refer to an LLO fragment that comprises a PEST sequence. In another embodiment, the terms refer to an LLO fragment that comprises a putative PEST sequence. In another embodiment, the terms refer to an LLO fragment that does not contain the activation domain at the carboxy terminus and does not include cysteine 484. In another embodiment, the terms refer to an LLO fragment that is not hemolytic. In another embodiment, the LLO fragment is rendered non-hemolytic by deletion or mutation of the activation domain. In another embodiment, the LLO fragment is rendered non-hemolytic by deletion or mutation of cysteine 484. In another embodiment, the LLO fragment is rendered non-hemolytic by deletion or mutation at another location. Each possibility represents a separate embodiment of the present invention. In another embodiment, the LLO fragment consists of about the first 441 AA of a wild-type LLO protein. In another embodiment, the LLO fragment consists of about the first 420 AA of LLO. In another embodiment, the LLO fragment is a non-hemolytic form of the LLO protein. In another embodiment, the LLO fragment contains residues of a homologous LLO protein that correspond to one of the above AA ranges. The residue numbers need not, in another embodiment, correspond exactly with the residue numbers enumerated above; e.g. if the homologous LLO protein has an insertion or deletion, relative to an LLO protein utilized herein, then the residue numbers can be adjusted accordingly. In another embodiment, the LLO fragment is any other LLO fragment known in the art. Each possibility represents a separate embodiment of the present invention. In another embodiment, the recombinant polypeptide of methods of the present invention is expressed by the recombinant In another embodiment, the recombinant In another embodiment, a plasmid comprised by a recombinant In another embodiment, a In another embodiment, a In another embodiment, the gene is phoP. In another embodiment, the gene is aroA. In another embodiment, the gene is aroC. In another embodiment, the gene is aroD. In another embodiment, the gene is plcB. In another embodiment, a In one embodiment, provided herein is a nucleic acid molecule that is used to transform the In one embodiment, a live attenuated It will be appreciated by a skilled artisan that the term “attenuation,” may encompass a diminution in the ability of the bacterium to cause disease in an animal. In other words, for example the pathogenic characteristics of the attenuated In yet another embodiment, a In one embodiment, the metabolic gene, the virulence gene, etc. is lacking in a chromosome of the In one embodiment, a recombinant It will be appreciated by a skilled artisan that the term “mutation” and grammatical equivalents thereof, include any type of mutation or modification to the sequence (nucleic acid or amino acid sequence), and includes a deletion mutation, a truncation, an inactivation, a disruption, or a translocation. These types of mutations are readily known in the art. In one embodiment, in order to select for auxotrophic bacteria comprising a plasmid encoding a metabolic enzyme or a complementing gene provided herein, transformed auxotrophic bacteria are grown on a media that will select for expression of the amino acid metabolism gene or the complementing gene. In another embodiment, a bacteria auxotrophic for D-glutamic acid synthesis is transformed with a plasmid comprising a gene for D-glutamic acid synthesis, and the auxotrophic bacteria will grow in the absence of D-glutamic acid, whereas auxotrophic bacteria that have not been transformed with the plasmid, or are not expressing the plasmid encoding a protein for D-glutamic acid synthesis, will not grow. In another embodiment, a bacterium auxotrophic for D-alanine synthesis will grow in the absence of D-alanine when transformed and expressing the plasmid of the present invention if the plasmid comprises an isolated nucleic acid encoding an amino acid metabolism enzyme for D-alanine synthesis. Such methods for making appropriate media comprising or lacking necessary growth factors, supplements, amino acids, vitamins, antibiotics, and the like are well known in the art, and are available commercially (Becton-Dickinson, Franklin Lakes, N.J.). In another embodiment, once the auxotrophic bacteria comprising the plasmid of the present invention have been selected on appropriate media, the bacteria are propagated in the presence of a selective pressure. Such propagation comprises growing the bacteria in media without the auxotrophic factor. The presence of the plasmid expressing an amino acid metabolism enzyme in the auxotrophic bacteria ensures that the plasmid will replicate along with the bacteria, thus continually selecting for bacteria harboring the plasmid. The skilled artisan, when equipped with the present disclosure and methods herein will be readily able to scale-up the production of the The skilled artisan will appreciate that, in another embodiment, other auxotroph strains and complementation systems are adopted for the use with this invention. In one embodiment, a recombinant In another embodiment, the dose is 1×107organisms. In another embodiment, the dose is 1×108organisms. In another embodiment, the dose is 1×109organisms. In another embodiment, the dose is 1.5×109organisms. In another embodiment, the dose is 2×109organisms. In another embodiment, the dose is 3×109organisms. In another embodiment, the dose is 4×109organisms. In another embodiment, the dose is 5×109organisms. In another embodiment, the dose is 6×109organisms. In another embodiment, the dose is 7×109organisms. In another embodiment, the dose is 8×109organisms. In another embodiment, the dose is 10×109organisms. In another embodiment, the dose is 1.5×1010organisms. In another embodiment, the dose is 2×1010organisms. In another embodiment, the dose is 2.5×1010organisms. In another embodiment, the dose is 3×1010organisms. In another embodiment, the dose is 3.3×1010organisms. In another embodiment, the dose is 4×1010organisms. In another embodiment, the dose is 5×1010organisms. Each dose and range of doses represents a separate embodiment of the present invention. In one embodiment, repeat administrations (doses) of compositions of this invention may be undertaken immediately following the first course of treatment or after an interval of days, weeks or months to achieve tumor regression. In another embodiment, repeat doses may be undertaken immediately following the first course of treatment or after an interval of days, weeks or months to achieve suppression of tumor growth. Assessment may be determined by any of the techniques known in the art, including diagnostic methods such as imaging techniques, analysis of serum tumor markers, biopsy, or the presence, absence or amelioration of tumor associated symptoms. It will be appreciated by the skilled artisan that the term “Boosting” may encompass administering an immunogenic composition or recombinant In one embodiment, a method of present invention further comprises the step of boosting the human subject with a recombinant In another embodiment, a method of present invention further comprises the step of inoculating the human subject with an immunogenic composition comprising the E7 antigen. In another embodiment, the immunogenic composition comprises a recombinant E7 protein or fragment thereof. In another embodiment, the immunogenic composition comprises a nucleotide molecule expressing a recombinant E7 protein or fragment thereof. In another embodiment, the non-Listerial inoculation is administered after the Listerial inoculation. In another embodiment, the non-Listerial inoculation is administered before the Listerial inoculation. Each possibility represents a separate embodiment of the present invention. The recombinant The present invention provides a number of Listerial species and strains for making or engineering an attenuated In another embodiment, a recombinant In another embodiment, the recombinant In another embodiment, the cell bank of methods and compositions of the present invention is a master cell bank. In another embodiment, the cell bank is a working cell bank. In another embodiment, the cell bank is Good Manufacturing Practice (GMP) cell bank. In another embodiment, the cell bank is intended for production of clinical-grade material. In another embodiment, the cell bank conforms to regulatory practices for human use. In another embodiment, the cell bank is any other type of cell bank known in the art. Each possibility represents a separate embodiment of the present invention. “Good Manufacturing Practices” are defined, in another embodiment, by (21 CFR 210-211) of the United States Code of Federal Regulations. In another embodiment, “Good Manufacturing Practices” are defined by other standards for production of clinical-grade material or for human consumption; e.g. standards of a country other than the United States. Each possibility represents a separate embodiment of the present invention. In another embodiment, a recombinant In another embodiment, a recombinant In another embodiment, a peptide of the present invention is a fusion peptide. In another embodiment, “fusion peptide” refers to a peptide or polypeptide comprising 2 or more proteins linked together by peptide bonds or other chemical bonds. In another embodiment, the proteins are linked together directly by a peptide or other chemical bond. In another embodiment, the proteins are linked together with 1 or more AA (e.g. a “spacer”) between the 2 or more proteins. Each possibility represents a separate embodiment of the present invention. In another embodiment, a vaccine of the present invention further comprises an adjuvant. The adjuvant utilized in methods and compositions of the present invention is, in another embodiment, a granulocyte/macrophage colony-stimulating factor (GM-CSF) protein. In another embodiment, the adjuvant comprises a GM-CSF protein. In another embodiment, the adjuvant is a nucleotide molecule encoding GM-CSF. In another embodiment, the adjuvant comprises a nucleotide molecule encoding GM-CSF. In another embodiment, the adjuvant is saponin QS21. In another embodiment, the adjuvant comprises saponin QS21. In another embodiment, the adjuvant is monophosphoryl lipid A. In another embodiment, the adjuvant comprises monophosphoryl lipid A. In another embodiment, the adjuvant is SBAS2. In another embodiment, the adjuvant comprises SBAS2. In another embodiment, the adjuvant is an unmethylated CpG-containing oligonucleotide. In another embodiment, the adjuvant comprises an unmethylated CpG-containing oligonucleotide. In another embodiment, the adjuvant is an immune-stimulating cytokine. In another embodiment, the adjuvant comprises an immune-stimulating cytokine. In another embodiment, the adjuvant is a nucleotide molecule encoding an immune-stimulating cytokine. In another embodiment, the adjuvant comprises a nucleotide molecule encoding an immune-stimulating cytokine. In another embodiment, the adjuvant is or comprises a quill glycoside. In another embodiment, the adjuvant is or comprises a bacterial mitogen. In another embodiment, the adjuvant is or comprises a bacterial toxin. In another embodiment, the adjuvant is or comprises any other adjuvant known in the art. Each possibility represents a separate embodiment of the present invention. In another embodiment, a nucleotide of the present invention is operably linked to a promoter/regulatory sequence that drives expression of the encoded peptide in the An N-terminal fragment of an ActA protein utilized in methods and compositions of the present invention has, in another embodiment, the sequence set forth in SEQ ID NO: 5. MRAMMVVFITANCITINPDIIFAATDSEDSSLNTDEWEEEKTEEQPSEVNTGPRYETAR EVSSRDIKELEKSNKVRNTNKADLIAMLKEKAEKGPNINNNNSEQTENAAINEEASG ADRPAIQVERRHPGLPSDSAAEIKKRRKAIASSDSELESLTYPDKPTKVNKKKVAKES VADASESDLDSSMQSADESSPQPLKANQQPFFPKVFKKIKDAGKWVRDKIDENPEVK KAIVDKSAGLIDQLLTKKKSEEVNASDFPPPPTDEELRLALPETPMLLGFNAPATSEPS SFEFPPPPTDEELRLALPETPMLLGFNAPATSEPSSFEFPPPPTEDELEHRETASSLDS SF TRGDLASLRNAINRHSQNFSDFPPIPTEEELNGRGGRP. In another embodiment, the ActA fragment comprises the sequence set forth in SEQ ID NO: 5. In another embodiment, the ActA fragment is any other ActA fragment known in the art. Each possibility represents a separate embodiment of the present invention. In another embodiment, the recombinant nucleotide encoding a fragment of an ActA protein comprises the sequence set forth in SEQ ID NO: 6: Atgcgtgcgatgatggtggttttcattactgccaattgcattacgattaaccccgacataatatttgcagcgacagatagcgaagattcta gtctaaacacagatgaatgggaagaagaaaaaacagaagagcaaccaagcgaggtaaatacgggaccaagatacgaaactgcacg tgaagtaagttcacgtgatattaaagaactagaaaaatcgaataaagtgagaaatacgaacaaagcagacctaatagcaatgttgaaag aaaaagcagaaaaaggtccaaatatcaataataacaacagtgaacaaactgagaatgcggctataaatgaagaggcttcaggagccg accgaccagctatacaagtggagcgtcgtcatccaggattgccatcggatagcgcagcggaaattaaaaaaagaaggaaagccatag catcatcggatagtgagatgaaagccttacttatccggataaaccaacaaaagtaaataagaaaaaagtggcgaaagagtcagttgcg gatgatctgaaagtgacttagattctagcatgcagtcagcagatgagtatcaccacaacctttaaaagcaaaccaacaaccattntccc taaagtatttaaaaaaataaaagatgcggggaaatgggtacgtgataaaatcgacgaaaatcctgaagtaaagaaagcgattgttgata aaagtgcagggttaattgaccaattattaaccaaaaagaaaagtgaagaggtaaatgatcggacttcccgccaccacctacggatgaa gagttaagacttgattgccagagacaccaatgatcttggttttaatgctcctgctacatcagaaccgagctcattcgaatttccaccacca cctacggatgaagagttaagacttgattgccagagacgccaatgatcttggttttaatgctcctgctacatcggaaccgagctcgttcga atttccaccgcctccaacagaagatgaactagaaatcatccgggaaacagcatcctcgctagattctagttttacaagaggggatttagct agtttgagaaatgctattaatcgccatagtcaaaatttctctgatttcccaccaatcccaacagaagaagagttgaacgggagaggcggt agacca. In another embodiment, the recombinant nucleotide has the sequence set forth in SEQ ID NO: 6. In another embodiment, the recombinant nucleotide comprises any other sequence that encodes a fragment of an ActA protein. Each possibility represents a separate embodiment of the present invention. In another embodiment of the methods and compositions of the present invention, a PEST amino acid AA sequence is fused to the E7 or E6 antigen. As provided herein, recombinant Thus, fusion of an antigen to other LM PEST amino acid sequences and PEST amino acid sequences derived from other prokaryotic organisms will also enhance immunogenicity of the antigen. The PEST amino acid AA sequence has, in another embodiment, a sequence selected from SEQ ID NO: 7-12. In another embodiment, the PEST amino acid sequence is a PEST amino acid sequence from the LM ActA protein. In another embodiment, the PEST amino acid sequence is KTEEQPSEVNTGPR (SEQ ID NO: 7), KASVTDTSEGDLDSSMQSADESTPQPLK (SEQ ID NO: 8), KNEEVNASDFPPPPTDEELR (SEQ ID NO: 9), or RGGIPTSEEFSSLNSGDFTDDENSETTEEEIDR (SEQ ID NO: 10). In another embodiment, the PEST amino acid sequence is from Streptolysin 0 protein of PEST amino acid sequences of other prokaryotic organism can be identified in accordance with methods such as described by, for example Rechsteiner and Rogers (1996, Trends Biochem. Sci. 21:267-271) for LM. Alternatively, PEST amino acid AA sequences from other prokaryotic organisms can also be identified based by this method. Other prokaryotic organisms wherein PEST amino acid AA sequences would be expected to include, but are not limited to, other In another embodiment, a PEST amino acid sequence is identified using any other method or algorithm known in the art, e.g the CaSPredictor (Garay-Malpartida H M, Occhiucci J M, Alves J, Belizario J E. Bioinformatics. 2005 June; 21 Suppl 1:i169-76). In another embodiment, the following method is used: A PEST index is calculated for each 30-35 AA stretch by assigning a value of 1 to the amino acids Ser, Thr, Pro, Glu, Asp, Asn, or Gln. The coefficient value (CV) for each of the PEST residue is 1 and for each of the other AA (non-PEST) is 0. Each method for identifying a PEST amino acid sequence represents a separate embodiment of the present invention. In another embodiment, the LLO protein, ActA protein, or fragment thereof of the present invention need not be that which is set forth exactly in the sequences set forth herein, but rather other alterations, modifications, or changes can be made that retain the functional characteristics of an LLO or ActA protein fused to an antigen as set forth elsewhere herein. In another embodiment, the present invention utilizes an analog of an LLO protein, ActA protein, or fragment thereof. Analogs differ, in another embodiment, from naturally occurring proteins or peptides by conservative AA sequence differences or by modifications which do not affect sequence, or by both. In another embodiment, either a whole E7 protein or a fragment thereof is fused to a LLO protein, ActA protein, or PEST amino acid sequence-containing peptide to generate a recombinant peptide of methods of the present invention. The E7 protein that is utilized (either whole or as the source of the fragments) has, in another embodiment, the sequence MHGDTPTLHEYMLDLQPETTDLYCYEQLNDSSEEEDEIDGPAGQAEPDRAHY NIVTFCCKCDSTLRLCVQSTHVDIRTLEDLLMGTLGIVCPICSQKP (SEQ ID No: 13). In another embodiment, the E7 protein is a homologue of SEQ ID No: 13. In another embodiment, the E7 protein is a variant of SEQ ID No: 13. In another embodiment, the E7 protein is an isomer of SEQ ID No: 13. In another embodiment, the E7 protein is a fragment of SEQ ID No: 13. In another embodiment, the E7 protein is a fragment of a homologue of SEQ ID No: 13. In another embodiment, the E7 protein is a fragment of a variant of SEQ ID No: 13. In another embodiment, the E7 protein is a fragment of an isomer of SEQ ID No: 13. Each possibility represents a separate embodiment of the present invention. In another embodiment, the sequence of the E7 protein is: MHGPKATLQDIVLHLEPQNEIPVDLLCHEQLSDSEEENDEIDGVNHQHLPARR AEPQRHTMLCMCCKCEARIELVVESSADDLRAFQQLFLNTLSFVCPWCASQQ (SEQ ID No: 14). In another embodiment, the E6 protein is a homologue of SEQ ID No: 14. In another embodiment, the E6 protein is a variant of SEQ ID No: 14. In another embodiment, the E6 protein is an isomer of SEQ ID No: 14. In another embodiment, the E6 protein is a fragment of SEQ ID No: 14. In another embodiment, the E6 protein is a fragment of a homologue of SEQ ID No: 14. In another embodiment, the E6 protein is a fragment of a variant of SEQ ID No: 14. In another embodiment, the E6 protein is a fragment of an isomer of SEQ ID No: 14. Each possibility represents a separate embodiment of the present invention. In another embodiment, the E7 protein has a sequence set forth in one of the following GenBank entries: M24215, NC_004500, V01116, X62843, or M14119. In another embodiment, the E7 protein is a homologue of a sequence from one of the above GenBank entries. In another embodiment, the E7 protein is a variant of a sequence from one of the above GenBank entries. In another embodiment, the E7 protein is an isomer of a sequence from one of the above GenBank entries. In another embodiment, the E7 protein is a fragment of a sequence from one of the above GenBank entries. In another embodiment, the E7 protein is a fragment of a homologue of a sequence from one of the above GenBank entries. In another embodiment, the E7 protein is a fragment of a variant of a sequence from one of the above GenBank entries. In another embodiment, the E7 protein is a fragment of an isomer of a sequence from one of the above GenBank entries. Each possibility represents a separate embodiment of the present invention. In another embodiment, either a whole E6 protein or a fragment thereof is fused to a LLO protein, ActA protein, or PEST amino acid sequence-containing peptide to generate a recombinant peptide of methods of the present invention. The E6 protein that is utilized (either whole or as the source of the fragments) has, in another embodiment, the sequence MHQKRTAMFQDPQERPRKLPQLCTELQTTIHDIILECVYCKQQLLRREVYDFA FRDLCIVYRDGNPYAVCDKCLKFYSKISEYRHYCYSLYGTTLEQQYNKPLCDLLIRCI NCQKPLCPEEKQRHLDKKQRFHNIRGRWTGRCMSCCRSSRTRRETQL (SEQ ID No: 15). In another embodiment, the E6 protein is a homologue of SEQ ID No: 15. In another embodiment, the E6 protein is a variant of SEQ ID No: 15. In another embodiment, the E6 protein is an isomer of SEQ ID No: 15. In another embodiment, the E6 protein is a fragment of SEQ ID No: 15. In another embodiment, the E6 protein is a fragment of a homologue of SEQ ID No: 15. In another embodiment, the E6 protein is a fragment of a variant of SEQ ID No: 15. In another embodiment, the E6 protein is a fragment of an isomer of SEQ ID No: 15. Each possibility represents a separate embodiment of the present invention. In another embodiment, the sequence of the E6 protein is: MARFEDPTRRPYKLPDLCTELNTSLQDIEITCVYCKTVLELTEVFEFAFKDLFV VYRDSIPHAACHKCIDFYSRIRELRHYSDSVYGDTLEKLTNTGLYNLLIRCLRCQKPL NPAEKLRHLNEKRRFHNIAGHYRGQCHSCCNRARQERLQRRRETQV (SEQ ID No: 16). In another embodiment, the E6 protein is a homologue of SEQ ID No: 16. In another embodiment, the E6 protein is a variant of SEQ ID No: 16. In another embodiment, the E6 protein is an isomer of SEQ ID No: 16. In another embodiment, the E6 protein is a fragment of SEQ ID No: 16. In another embodiment, the E6 protein is a fragment of a homologue of SEQ ID No: 16. In another embodiment, the E6 protein is a fragment of a variant of SEQ ID No: 16. In another embodiment, the E6 protein is a fragment of an isomer of SEQ ID No: 16. Each possibility represents a separate embodiment of the present invention. In another embodiment, the E6 protein has a sequence set forth in one of the following GenBank entries: M24215, M14119, NC_004500, V01116, X62843, or M14119. In another embodiment, the E6 protein is a homologue of a sequence from one of the above GenBank entries. In another embodiment, the E6 protein is a variant of a sequence from one of the above GenBank entries. In another embodiment, the E6 protein is an isomer of a sequence from one of the above GenBank entries. In another embodiment, the E6 protein is a fragment of a sequence from one of the above GenBank entries. In another embodiment, the E6 protein is a fragment of a homologue of a sequence from one of the above GenBank entries. In another embodiment, the E6 protein is a fragment of a variant of a sequence from one of the above GenBank entries. In another embodiment, the E6 protein is a fragment of an isomer of a sequence from one of the above GenBank entries. Each possibility represents a separate embodiment of the present invention. In another embodiment, “homology” refers to identity to an LLO sequence (e.g. to one of SEQ ID No: 2-4) of greater than 60%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 64%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 68%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 72%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 75%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 78%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 80%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 82%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 83%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 85%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 87%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 88%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 90%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 92%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 93%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 95%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 96%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 97%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 98%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of greater than 99%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 2-4 of 100%. Each possibility represents a separate embodiment of the present invention. In another embodiment, “homology” refers to identity to an E7 sequence (e.g. to one of SEQ ID No: 13-14) of greater than 60%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 62%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 64%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 68%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 72%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 75%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 78%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 80%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 82%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 83%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 85%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 87%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 88%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 90%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 92%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 93%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 95%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 96%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 97%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 98%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of greater than 99%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 13-14 of 100%. Each possibility represents a separate embodiment of the present invention. In another embodiment, “homology” refers to identity to an E6 sequence (e.g. to one of SEQ ID No: 15-16) of greater than 60%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 64%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 68%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 72%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 75%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 78%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 80%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 82%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 83%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 85%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 87%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 88%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 90%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 92%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 93%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 95%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 96%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 97%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 98%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of greater than 99%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 15-16 of 100%. Each possibility represents a separate embodiment of the present invention. In another embodiment, “homology” refers to identity to a PEST amino acid sequence (e.g. to one of SEQ ID No: 1, and 7-12) or to an ActA sequence (e.g. to one of SEQ ID No: 5-6) of greater than 60%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 60%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 64%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 68%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 72%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 75%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 78%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 80%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 82%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 83%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 85%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 87%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 88%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 90%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 92%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 93%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 95%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 96%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 97%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 98%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of greater than 99%. In another embodiment, “homology” refers to identity to one of SEQ ID No: 1, and 7-12 or SEQ ID No: 5-6 of 100%. Each possibility represents a separate embodiment of the present invention. Protein and/or peptide homology for any AA sequence listed herein is determined, in one embodiment, by methods well described in the art, including immunoblot analysis, or via computer algorithm analysis of AA sequences, utilizing any of a number of software packages available, via established methods. Some of these packages include the FASTA, BLAST, MPsrch or Scanps packages, and employ, in other embodiments, the use of the Smith and Waterman algorithms, and/or global/local or BLOCKS alignments for analysis, for example. Each method of determining homology represents a separate embodiment of the present invention. In another embodiment, the LLO protein, ActA protein, or fragment thereof is attached to the antigen by chemical conjugation. In another embodiment, glutaraldehyde is used for the conjugation. In another embodiment, the conjugation is performed using any suitable method known in the art. Each possibility represents another embodiment of the present invention. In another embodiment, fusion proteins of the present invention are prepared by any suitable method, including, for example, cloning and restriction of appropriate sequences or direct chemical synthesis by methods discussed below. In another embodiment, subsequences are cloned and the appropriate subsequences cleaved using appropriate restriction enzymes. The fragments are then ligated, in another embodiment, to produce the desired DNA sequence. In another embodiment, DNA encoding the fusion protein is produced using DNA amplification methods, for example polymerase chain reaction (PCR). First, the segments of the native DNA on either side of the new terminus are amplified separately. The 5 end of the one amplified sequence encodes the peptide linker, while the 3′ end of the other amplified sequence also encodes the peptide linker. Since the 5′ end of the first fragment is complementary to the 3′ end of the second fragment, the two fragments (after partial purification, e.g. on LMP agarose) can be used as an overlapping template in a third PCR reaction. The amplified sequence will contain codons, the segment on the carboxy side of the opening site (now forming the amino sequence), the linker, and the sequence on the amino side of the opening site (now forming the carboxyl sequence). The insert is then ligated into a plasmid. In another embodiment, the LLO protein, ActA protein, or fragment thereof and the antigen, or fragment thereof are conjugated by a means known to those of skill in the art. In another embodiment, the antigen, or fragment thereof is conjugated, either directly or through a linker (spacer), to the ActA protein or LLO protein. In another embodiment, the chimeric molecule is recombinantly expressed as a single-chain fusion protein. In another embodiment, a fusion peptide of the present invention is synthesized using standard chemical peptide synthesis techniques. In another embodiment, the chimeric molecule is synthesized as a single contiguous polypeptide. In another embodiment, the LLO protein, ActA protein, or fragment thereof; and the antigen, or fragment thereof are synthesized separately, then fused by condensation of the amino terminus of one molecule with the carboxyl terminus of the other molecule, thereby forming a peptide bond. In another embodiment, the ActA protein or LLO protein and antigen are each condensed with one end of a peptide spacer molecule, thereby forming a contiguous fusion protein. In another embodiment, the peptides and proteins of the present invention are prepared by solid-phase peptide synthesis (SPPS) as described by Stewart et al. in Solid Phase Peptide Synthesis, 2nd Edition, 1984, Pierce Chemical Company, Rockford, Ill.; or as described by Bodanszky and Bodanszky (The Practice of Peptide Synthesis, 1984, Springer-Verlag, New York). In another embodiment, a suitably protected AA residue is attached through its carboxyl group to a derivatized, insoluble polymeric support, such as cross-linked polystyrene or polyamide resin. “Suitably protected” refers to the presence of protecting groups on both the alpha-amino group of the amino acid, and on any side chain functional groups. Side chain protecting groups are generally stable to the solvents, reagents and reaction conditions used throughout the synthesis, and are removable under conditions which will not affect the final peptide product. Stepwise synthesis of the oligopeptide is carried out by the removal of the N-protecting group from the initial AA, and couple thereto of the carboxyl end of the next AA in the sequence of the desired peptide. This AA is also suitably protected. The carboxyl of the incoming AA can be activated to react with the N-terminus of the support-bound AA by formation into a reactive group such as formation into a carbodiimide, a symmetric acid anhydride or an “active ester” group such as hydroxybenzotriazole or pentafluorophenly esters. The pharmaceutical compositions containing vaccines and compositions of the present invention are, in another embodiment, administered to a subject by any method known to a person skilled in the art, such as parenterally, paracancerally, transmucosally, transdermally, intramuscularly, intravenously, intra-dermally, subcutaneously, intra-peritonealy, intra-ventricularly, intra-cranially, intra-vaginally or intra-tumorally. In another embodiment of the methods and compositions provided herein, the vaccines or compositions are administered orally, and are thus formulated in a form suitable for oral administration, i.e. as a solid or a liquid preparation. Suitable solid oral formulations include tablets, capsules, pills, granules, pellets and the like. Suitable liquid oral formulations include solutions, suspensions, dispersions, emulsions, oils and the like. In another embodiment of the present invention, the active ingredient is formulated in a capsule. In accordance with this embodiment, the compositions of the present invention comprise, in addition to the active compound and the inert carrier or diluent, a hard gelating capsule. In another embodiment, the vaccines or compositions are administered by intravenous, intra-arterial, or intra-muscular injection of a liquid preparation. Suitable liquid formulations include solutions, suspensions, dispersions, emulsions, oils and the like. In one embodiment, the pharmaceutical compositions are administered intravenously and are thus formulated in a form suitable for intravenous administration. In another embodiment, the pharmaceutical compositions are administered intra-arterially and are thus formulated in a form suitable for intra-arterial administration. In another embodiment, the pharmaceutical compositions are administered intra-muscularly and are thus formulated in a form suitable for intra-muscular administration. It will be appreciated by a skilled artisan that the term “treating” may encompass both therapeutic treatment and prophylactic or preventative measures, wherein the object is to prevent or lessen the targeted pathologic condition or disorder as described herein. Thus, in one embodiment, treating may include directly affecting or curing, suppressing, inhibiting, preventing, reducing the severity of, delaying the onset of, reducing symptoms associated with the disease, disorder or condition, or a combination thereof. Thus, in one embodiment, “treating” may encompass inter alia delaying progression, expediting remission, inducing remission, augmenting remission, speeding recovery, increasing efficacy of or decreasing resistance to alternative therapeutics, or a combination thereof. In one embodiment, “preventing” or “impeding” may encompass, inter alia, delaying the onset of symptoms, preventing relapse to a disease, decreasing the number or frequency of relapse episodes, increasing latency between symptomatic episodes, or a combination thereof. In one embodiment, “suppressing” or “inhibiting”, may encompass, inter alia, reducing the severity of symptoms, reducing the severity of an acute episode, reducing the number of symptoms, reducing the incidence of disease-related symptoms, reducing the latency of symptoms, ameliorating symptoms, reducing secondary symptoms, reducing secondary infections, prolonging patient survival, or a combination thereof. In one embodiment, symptoms are primary, while in another embodiment, symptoms are secondary. In one embodiment, “primary” refers to a symptom that is a direct result of a particular disease or disorder, while in one embodiment, “secondary” refers to a symptom that is derived from or consequent to a primary cause. In one embodiment, the compounds for use in the present invention treat primary or secondary symptoms or secondary complications. In another embodiment, “symptoms” may be any manifestation of a disease or pathological condition. In another embodiment, the present invention provides a kit comprising vaccine of the present invention, an applicator, and instructional material that describes use of the methods of the invention. Although model kits are described below, the contents of other useful kits will be apparent to the skilled artisan in light of the present disclosure. Each of these kits represents a separate embodiment of the present invention. In one embodiment, the singular forms of words such as “a,” “an,” and “the,” include their corresponding plural references unless the context clearly dictates otherwise. Throughout this application, various embodiments of this invention may be presented in a range format. It should be understood that the description in range format is merely for convenience and brevity and should not be construed as an inflexible limitation on the scope of the invention. Accordingly, the description of a range should be considered to have specifically disclosed all the possible sub ranges as well as individual numerical values within that range. For example, description of a range such as from 1 to 6 should be considered to have specifically disclosed sub ranges such as from 1 to 3, from 1 to 4, from 1 to 5, from 2 to 4, from 2 to 6, from 3 to 6 etc., as well as individual numbers within that range, for example, 1, 2, 3, 4, 5, and 6. This applies regardless of the breadth of the range. Whenever a numerical range is indicated herein, it is meant to include any cited numeral (fractional or integral) within the indicated range. The phrases “ranging/ranges between” a first indicate number and a second indicate number and “ranging/ranges from” a first indicate number “to” a second indicate number are used herein interchangeably and are meant to include the first and second indicated numbers and all the fractional and integral numerals there between. It will be appreciated by a skilled artisan that the term “about” when used to modify a numerically defined parameter may encompass variation of the parameter in quantitative terms plus or minus 5%, or in another embodiment plus or minus 10%, or in another embodiment plus or minus 15%, or in another embodiment plus or minus 20% of stated numerical value for that parameter. It is to be understood by the skilled artisan that the term “subject” can encompass a mammal including an adult human or a human child, teenager or adolescent in need of therapy for, or susceptible to, a condition or its sequelae, and also may include non-human mammals such as dogs, cats, pigs, cows, sheep, goats, horses, rats, and mice. It will also be appreciated that the term may encompass livestock. The term “subject” does not exclude an individual that is normal in all respects. It will be appreciated by the skilled artisan that the term “mammal” for purposes of treatment refers to any animal classified as a mammal, including, but not limited to, humans, domestic and farm animals, and zoo, sports, or pet animals, such as canines, including dogs, and horses, cats, cattle, pigs, sheep, etc. In the following examples, numerous specific details are set forth in order to provide a thorough understanding of the invention. However, it will be understood by those skilled in the art that the present invention may be practiced without these specific details. In other instances, well-known methods, procedures, and components have not been described in detail so as not to obscure the present invention. Thus these examples should in no way be construed, as limiting the broad scope of the invention. The C57BL/6 syngeneic TC-1 tumor was immortalized with HPV-16 E6 and E7 and transformed with the c-Ha-ras oncogene. TC-1, provided by T. C. Wu (Johns Hopkins University School of Medicine, Baltimore, Md.) is a highly tumorigenic lung epithelial cell expressing low levels of with HPV-16 E6 and E7 and transformed with the c-Ha-ras oncogene. TC-1 was grown in RPMI 1640, 10% FCS, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 100 μM nonessential amino acids, 1 mM sodium pyruvate, 50 micromolar (mcM) 2-ME, 400 microgram (mcg)/ml G418, and 10% National Collection Type Culture-109 medium at 37° with 10% CO2. C3 is a mouse embryo cell from C57BL/6 mice immortalized with the complete genome of HPV 16 and transformed with pEJ-ras. EL-4/E7 is the thymoma EL-4 retrovirally transduced with E7. Tumors were measured every other day with calipers spanning the shortest and longest surface diameters. The mean of these two measurements was plotted as the mean tumor diameter in millimeters against various time points. Mice were sacrificed when the tumor diameter reached 20 mm Tumor measurements for each time point are shown only for surviving mice. Six- to 8-wk-old C57BL/6 mice (Charles River) received 2×105TC-1 cells s.c. on the left flank. One week following tumor inoculation, the tumors had reached a palpable size of 4-5 mm in diameter. Groups of eight mice were then treated with 0.1 LD50i.p. Lm-LLO-E7 (107CFU), Lm-E7 (106CFU), Lm-LLO-NP (107CFU), or Lm-Gag (5×105CFU) on days 7 and 14. C57BL/6 mice, 6-8 wk old, were immunized i.p. with 0.1LD50Lm-LLO-E7, Lm-E7, Lm-LLO-NP, or Lm-Gag. Ten days post-immunization, spleens were harvested. Splenocytes were established in culture with irradiated TC-1 cells (100:1, splenocytes:TC-1) as feeder cells; stimulated in vitro for 5 days, then used in a standard51Cr release assay, using the following targets: EL-4, EL-4/E7, or EL-4 pulsed with E7 H-2b peptide (RAHYNIVTF). E:T cell ratios, performed in triplicate, were 80:1, 40:1, 20:1, 10:1, 5:1, and 2.5:1. Following a 4-h incubation at 37° C., cells were pelleted, and 50 μl supernatant was removed from each well. Samples were assayed with a Wallac 1450 scintillation counter (Gaithersburg, Md.). The percent specific lysis was determined as [(experimental counts per minute (cpm)−spontaneous cpm)/(total cpm−spontaneous cpm)]×100. C57BL/6 mice were immunized with 0.1 LD50and boosted by i.p. injection 20 days later with 1 LD50Lm-LLO-E7, Lm-E7, Lm-LLO-NP, or Lm-Gag. Six days after boosting, spleens were harvested from immunized and naive mice. Splenocytes were established in culture at 5×105/well in flat-bottom 96-well plates with 2.5×104, 1.25×104, 6×103, or 3×103irradiated TC-1 cells/well as a source of E7 Ag, or without TC-1 cells or with 10 μg/ml Con A. Cells were pulsed 45 h later with 0.5 μCi [3H]thymidine/well. Plates were harvested 18 h later using a Tomtec harvester 96 (Orange, Conn.), and proliferation was assessed with a Wallac 1450 scintillation counter. The change in cpm was calculated as experimental cpm—no Ag cpm. C57BL/6 mice were immunized intravenously (i.v.) with 0.1 LD50Lm-LLO-E7 or Lm-E7 and boosted 30 days later. Three-color flow cytometry for CD8 (53-6.7, PE conjugated), CD62 ligand (CD62L; MEL-14, APC conjugated), and E7 H-2Db tetramer was performed using a FACSCalibur® flow cytometer with CellQuest® software (Becton Dickinson, Mountain View, Calif.). Splenocytes harvested 5 days after the boost were stained at room temperature (rt) with H-2Db tetramers loaded with the E7 peptide (RAHYNIVTF) or a control (HIV-Gag) peptide. Tetramers were used at a 1/200 dilution and were provided by Dr. Larry R. Pease (Mayo Clinic, Rochester, Minn.) and by the NIAID Tetramer Core Facility and the NIH AIDS Research and Reference Reagent Program. Tetramer, CD8, CD62Llowcells were analyzed. 24 C57BL/6 mice were inoculated with 5×105B16F0-Ova cells. On days 3, 10 and 17, groups of 8 mice were immunized with 0.1 LD50Lm-OVA (106cfu), Lm-LLO-OVA (108cfu) and eight animals were left untreated. For comparisons of tumor diameters, mean and SD of tumor size for each group were determined, and statistical significance was determined by Student's t test. p≦0.05 was considered significant. Lm-E7 and Lm-LLO-E7 were compared for their abilities to impact on TC-1 growth. Subcutaneous tumors were established on the left flank of C57BL/6 mice. Seven days later tumors had reached a palpable size (4-5 mm). Mice were vaccinated on days 7 and 14 with 0.1 LD50Lm-E7, Lm-LLO-E7, or, as controls, Lm-Gag and Lm-LLO-NP. Lm-LLO-E7 induced complete regression of 75% of established TC-1 tumors, while tumor growth was controlled in the other 2 mice in the group ( In other experiments, similar results were obtained with 2 other E7-expressing tumor cell lines: C3 and EL-4/E7. To confirm the efficacy of vaccination with Lm-LLO-E7, animals that had eliminated their tumors were re-challenged with TC-1 or EL-4/E7 tumor cells on day 60 or day 40, respectively. Animals immunized with Lm-LLO-E7 remained tumor free until termination of the experiment (day 124 in the case of TC-1 and day 54 for EL-4/E7). Thus, expression of an antigen as a fusion protein with ΔLLO enhances the immunogenicity of the antigen. To measure induction of T cells by Lm-E7 with Lm-LLO-E7, TC-1-specific proliferative responses, a measure of antigen-specific immunocompetence, were measured in immunized mice. Splenocytes from Lm-LLO-E7-immunized mice proliferated when exposed to irradiated TC-1 cells as a source of E7, at splenocyte: TC-1 ratios of 20:1, 40:1, 80:1, and 160:1 ( 500 mcl (microliter) of MATRIGEL®, comprising 100 mcl of 2×105TC-1 tumor cells in phosphate buffered saline (PBS) plus 400 mcl of MATRIGEL® (BD Biosciences, Franklin Lakes, N.J.) were implanted subcutaneously on the left flank of 12 C57BL/6 mice (n=3). Mice were immunized intraperitoneally on day 7, 14 and 21, and spleens and tumors were harvested on day 28. Tumor MATRIGELs were removed from the mice and incubated at 4° C. overnight in tubes containing 2 milliliters (ml) of RP 10 medium on ice. Tumors were minced with forceps, cut into 2 mm blocks, and incubated at 37° C. for 1 hour with 3 ml of enzyme mixture (0.2 mg/ml collagenase-P, 1 mg/ml DNAse-1 in PBS). The tissue suspension was filtered through nylon mesh and washed with 5% fetal bovine serum+0.05% of NaN3in PBS for tetramer and IFN-gamma staining. Splenocytes and tumor cells were incubated with 1 micromole (mcm) E7 peptide for 5 hours in the presence of brefeldin A at 107cells/ml. Cells were washed twice and incubated in 50 mcl of anti-mouse Fc receptor supernatant (2.4 G2) for 1 hour or overnight at 4° C. Cells were stained for surface molecules CD8 and CD62L, permeabilized, fixed using the permeabilization kit Golgi-Stop® or Golgi-Plug® (Pharmingen, San Diego, Calif.), and stained for IFN-gamma. 500,000 events were acquired using two-laser flow cytometer FACSCalibur and analyzed using Cellquest Software (Becton Dickinson, Franklin Lakes, N.J.). Percentages of IFN-gamma secreting cells within the activated (CD62Llow) CD8+ T cells were calculated. For tetramer staining, H-2Dbtetramer was loaded with phycoerythrin (PE)-conjugated E7 peptide (RAHYNIVTF, SEQ ID NO: 24), stained at rt for 1 hour, and stained with anti-allophycocyanin (APC) conjugated MEL-14 (CD62L) and FITC-conjugated CD8+ at 4° C. for 30 mm Cells were analyzed comparing tetramer+CD8+ CD62Llowcells in the spleen and in the tumor. To analyze the ability of Lm-ActA-E7 to enhance antigen specific immunity, mice were implanted with TC-1 tumor cells and immunized with either Lm-LLO-E7 (1×107CFU), Lm-E7 (1×106CFU), or Lm-ActA-E7 (2×108CFU), or were untreated (naïve). Tumors of mice from the Lm-LLO-E7 and Lm-ActA-E7 groups contained a higher percentage of IFN-gamma-secreting CD8+ T cells ( In another experiment, tumor-bearing mice were administered Lm-LLO-E7, Lm-PEST-E7, Lm-ΔPEST-E7, or Lm-E7epi, and levels of E7-specific lymphocytes within the tumor were measured. Mice were treated on days 7 and 14 with 0.1 LD50of the 4 vaccines. Tumors were harvested on day 21 and stained with antibodies to CD62L, CD8, and with the E7/Db tetramer. An increased percentage of tetramer-positive lymphocytes within the tumor were seen in mice vaccinated with Lm-LLO-E7 and Lm-PEST-E7 ( Thus, Lm-LLO-E7, Lm-ActA-E7, and Lm-PEST-E7 are each efficacious at induction of tumor-infiltrating CD8+ T cells and tumor regression. Bacterial Strains Bacteria from a single clone expressing the passenger antigen and/or fusion protein were selected and cultured in BHI broth overnight. Aliquots of this culture were frozen at −70° C. with no additives. From this stock, cultures were grown to 0.1-0.2 O.D. at 600 nm, and aliquots were again frozen at −70° C. with no additives. To prepare cloned bacterial pools, the above procedure was used, but after each passage a number of bacterial clones were selected and checked for expression of the target antigen, as described herein. Clones in which expression of the foreign antigen was confirmed were used for the next passage. 6-8 week old female BALB/c (H-2d) mice were purchased from Jackson Laboratories (Bar Harbor, Me.) and were maintained in a pathogen-free microisolator environment. The titer of viable bacteria in an aliquot of stock culture, stored frozen at −70° C., was determined by plating on BHI agar plates on thawing and prior to use. In all, 5×105bacteria were injected intravenously into BALB/c mice. After 3 days, spleens were harvested, homogenized, and serial dilutions of the spleen homogenate were incubated in BHI broth overnight and plated on BHI agar plates. For further passage, aliquots were again grown to 0.1-0.2 O.D., frozen at −70° C., and bacterial titer was again determined by serial dilution. After the initial passage (passage 0), this sequence was repeated for a total of 4 times. Lymphocytes were cultured for 5 hours in complete RPMI-10 medium supplemented with 50 U/ml human recombinant IL-2 and 1 microliter/ml Brefeldin A (Golgistop™; PharMingen, San Diego, Calif.) in the presence or absence of either the cytotoxic T-cell (CTL) epitope for HIV-GAG (AMQMLKETI; SEQ ID No: 25), Three different constructs were used to determine the impact of passaging on recombinant Passaging the bacteria resulted in an increase in bacterial virulence, as measured by numbers of surviving bacteria in the spleen, with each of the first 2 passages. For Lm-Gag and Lm-LLO-E7, virulence increased with each passage up to passage 2 ( Thus, passage through mice increases the virulence of Passaging Increases the Ability of Next, the effect of passaging on induction of antigen-specific CD8+ T cells was determined by intracellular cytokine staining with immunodominant peptides specific for MHC-class I using HIV-Gag peptide AMQMLKETI (SEQ ID No: 25) and LLO 91-99 (GYKDGNEYI; SEQ ID No: 26). Injection of 103CFU passaged bacteria (Lm-Gag) into mice elicited significant numbers of HIV-Gag-specific CD8+ T cells, while the same dose of non-passaged Lm-Gag induced no detectable Gag-specific CD8+ T cells. Even increasing the dose of unpassaged bacteria 100-fold did not compensate for their relative avirulence; in fact, no detectable Gag-specific CD8+ T cells were elicited even at the higher dose. The same dose increase with passaged bacteria increased Gag-specific T cell induction by 50% ( Thus, passage through mice increases the immunogenicity of Development of Protocol for Plasmid Extraction from 1 mL of Seven 2.5 mL samples of the culture were pelleted (15000 rpm for 5 minutes), and pellets were incubated at 37° C. with 50 μl lysozyme solution for varying amounts of time, from 0-60 minutes. Lysozyme solution:
After incubation with the lysozyme, the suspensions were centrifuged as before and the supernatants discarded. Each pellet was then subjected to plasmid extraction by a modified version of the QIAprep Spin Miniprep Kit® (Qiagen, Germantown, Md.) protocol. The changes to the protocol were as follows:
In other experiments, the cells were incubated for 15 min in P1 buffer+Lysozyme, then incubated with P2 (lysis buffer) and P3 (neutraliztion buffer) at room temperature. Equal volumes of the isolated plasmid DNA from each subculture were run on an 0.8% agarose gel stained with ethidium bromide and visualized for any signs of structural or segregation instability. The results showed that plasmid extraction from These results provide an effective method for plasmid extraction from Dilutions of the original culture were plated onto plates containing LB or TB agar in the absence or presence of 34 μg/mL CAP. The differences between the counts on selective and non-selective agar were used to determine whether there was any gross segregational instability of the plasmid. The genetic stability (i.e. the extent to which the plasmid is retained by or remains stably associated with the bacteria in the absence of selection pressure; e.g. antibiotic selection pressure) of the pGG55 plasmid in Plasmid stability was also monitored during the stability study by replica plating on agar plates at each stage of the subculture. Consistent with the results from the agarose gel electrophoresis, there was no overall change in the number of plasmid-containing cells throughout the study in either LB or TB liquid culture ( These findings demonstrate that prfA-encoding plasmids exhibit stability in the absence of antibiotic in The primers used for amplification of the prfA gene and discrimination of the D133V mutation are shown in Table 1. Stock solutions of the primers ADV451, 452 and 453 were prepared by diluting the primers in TE buffer to 400 μM. An aliquot of the stock solution was further diluted to 20 μM in water (PCR grade) to prepare a working solution. Primers were stored at −20° C. The reagents used in the PCR are shown in Table 2. pGG55 plasmids with (pGG55 D133V) and without (pGG55 WT) the prfA mutation were extracted and purified by midipreparations either from prfA Specific PCR Protocol to Test Clinical Grade Material The reaction mixture contained 1×PCR buffer, 1.5 mM MgCl2, 0.8 mM dNTPs, 0.4 μM of each primer, 0.05 U/μl of Taq DNA polymerase and 0.04 ng/μl of the pGG55 D133V template plasmid. For each test, 10 tubes were required and the key components in each tube in a 25 μl reaction are shown in the Table 3. For the PCR reaction, a master mix was prepared with enough reagents for 11 reactions as shown in Table 4, and 24 μl of this PCR mix was added to each tube. Subsequently, a total of 1 μl of the serially diluted pGG55 WT plasmid was added to the corresponding tubes: 1 ng in tube 3; 100 pg in tube 4; 10 pg in tube 5; 1 pg in tube 6; 100 fg in tube 7; 10 fg in tube 8; 1 fg in tube 9; 0.1 fg in tube 10. This serial dilution was used to calibrate a standard curve to determine the method sensitivity. Additionally, 0.5 μl of water and 0.5 μl of primer ADV451 (20 μM stock) were added in tube 1, and 1 μl of water added in tube 2, completing 25 μl of final volume. The quantities of each reagent per tube for a 25 μl reaction are shown in Table 5. The PCR cycling conditions used in the reaction are shown in Table 6. After conclusion of the PCR reaction, 5 μl of gel-loading buffer (6×, with bromophenol blue) was added to each sample and 10 μl were analyzed by electrophoresis in 1.2% agarose gel in TBE buffer. The gel dimensions were 7 cm×7 cm×1 cm with a 15 sample wells (1 mm×2 mm) comb. The gel was run at 100 V for ˜30 minutes, until the bromophenol blue dye reached the middle of the gel. The gel was stained in ethidium bromide (0.5 μg/ml) for 20 minutes, destaining in water for 10 minutes. The gel is visualized by illumination with UV light and photographed. The image was analyzed using a band densitometry software (Quantity One version 4.5.1, BioRad). Sequencing of the plasmids was done using the dideoxy sequencing method. The plasmids pGG55 D133V and pGG55 WT were mixed at different ratios (1:1, 1:10, 1;100, 1:1,000 and 1:10,000). The total amount of plasmid in the mixture was kept constant (500 μg) and the plasmid containing the wild-type sequence was 10-fold serially diluted in relation to the D133V plasmid to determine the sensitivity of the method. To estimate the sensitivity of sequencing in detecting the wild-type prfA sequence, the pGG55 D133V and WT plasmids were mixed at the different ratios and sequenced. The results are shown in Given the low sensitivity of sequencing to detect rare events, it became imperative to develop a more sensitive method with similar specificity to detect reversion of the D133V mutation to wild-type. To achieve this goal, we designed a PCR-based method that specifically amplifies the wild-type sequence and is sensitive enough to detect at least 1 wild-type copy of prfA in 10,000,000 copies of the D133V mutated sequence. We designed 3 primers for this method: ADV451, ADV452 and ADV453 (Table 1). Both ADV451 and ADV452 are forward primers and differ in the last nucleotide at the 3′ position to discriminate the A→T (D133V) mutation at position 398 of the prfA gene. The ADV453 primer is the reverse primer located approximately 300 bp downstream the annealing site of the ADV451 and ADV452 primers ( The reaction using the primer ADV451 was very specific and amplified the mutated D133V prfA sequence (lanes 1 to 3), but not the wild-type sequence (lanes 4 to 6). However, a very faint band can be detected in lane 4, when 5 ng of template DNA was used, but not with 1 ng ( As shown in The sensitivity of the reaction was tested using 1 ng of template DNA. For the plasmid carrying the wild-type pifA sequence, decreasing amounts of DNA (corresponding to 10-fold dilutions from 10−1to 10−7), were also included in the reaction to estimate the sensitivity. In these reactions only the primers ADV452 and ADV453 were used. In a PCR reaction with 30 cycles (10 cycles with annealing temperature of 53° C. and an additional 20 cycles with annealing temperature of 50° C.), the sensitivity of the method was 1 in 100,000 (data not shown). As shown in This strain is approx. 4-5 logs more attenuated than the wild-type parent strain 10403S and secretes the fusion protein tLLO-E7. This immunotherapy is based on the backbone XFL7, which is derived from 10403S by the irreversible deletion in the virulence gene transcription activator prfA. PrfA regulates the transcription of several virulence genes such as Listeriolysin O (LLO), ActA, PlcA (phospholipase A), PlcB (phospholipase B) etc that are required for in vivo intracellular growth and survival of While certain features of the invention have been illustrated and described herein, many modifications, substitutions, changes, and equivalents will now occur to those of ordinary skill in the art. It is, therefore, to be understood that the appended claims are intended to cover all such modifications and changes as fall within the true spirit of the invention. The present invention provides methods of treating, protecting against and inducing an immune response against a tumor or cancer, comprising the step of administering to a subject a recombinant Listeria strain. In one embodiment the present invention relates to a recombinant Listeria strain, said recombinant Listeria strain comprising a recombinant nucleic add, said nucleic add comprising a first open reading frame encoding a recombinant polypeptide comprising a first N-terminal fragment of an LLO protein fused to a heterologous antigen or fragment thereof, and wherein said recombinant nucleic add further comprises a second open reading frame encoding a mutant PrfA protein. 1. A recombinant 2. The recombinant 3. The recombinant 4. The recombinant 5. The recombinant 6. The recombinant 7. The recombinant 8. The recombinant 9. The recombinant 10. The recombinant 11. The recombinant 12. A pharmaceutical composition comprising the recombinant 13. A method of inducing an immune response against a tumor or a cancer in a human subject, the method comprising the step of administering to said subject a recombinant 14. The recombinant 15. The method of any one of 16. The method 17. The method of any one of 18. The method of any one of 19. The method of any one of 20. The method of any one of 21. The method of 22. The method of any one of 23. The method of any one of 24. The method of any one of 25. The method of any one of 26. The method of any one of 27. The method of any one of 28. The method of any one of 29. The method of any one of 30. The method of any one of 31. The method of any one of 32. The method of FIELD OF INVENTION
BACKGROUND OF THE INVENTION
SUMMARY OF THE INVENTION
BRIEF DESCRIPTION OF THE DRAWINGS
DETAILED DESCRIPTION OF THE INVENTION
(SEQ ID NO: 31) 1 atgaacgctc aagcagaaga attcaaaaaa tatttagaaa ctaacgggat aaaaccaaaa 61 caatttcata aaaaagaact tatttttaac caatgggatc cacaagaata ttgtattttt 121 ctatatgatg gtatcacaaa gctcacgagt attagcgaga acgggaccat catgaattta 181 caatactaca aaggggcttt cgttataatg tctggcttta ttgatacaga aacatcggtt 241 ggctattata atttagaagt cattagcgag caggctaccg catacgttat caaaataaac 301 gaactaaaag aactactgag caaaaatctt acgcactttt tctatgtttt ccaaacccta 361 caaaaacaag tttcatacag cctagctaaa tttaatgatt tttcgattaa cgggaagctt 421 ggctctattt gcggtcaact tttaatcctg acctatgtgt atggtaaaga aactcctgat 481 ggcatcaaga ttacactgga taatttaaca atgcaggagt taggatattc aagtggcatc 541 gcacatagct cagctgttag cagaattatt tccaaattaa agcaagagaa agttatcgtg 601 tataaaaatt catgctttta tgtacaaaat cttgattatc tcaaaagata tgcccctaaa 661 ttagatgaat ggttttattt agcatgtcct gctacttggg gaaaattaaa ttaa (SEQ ID NO: 32) M N A Q A E E F K K Y L E T N G I K P K Q F H K K E L I F N Q W D P Q E Y C I F L Y D G I T K L T S I S E N G T I M N L Q Y Y K G A F V I M S G F I D T E T S V G Y Y N L E V I S E Q A T A Y V I K I N E L K E L L S K N L T H F F Y V F Q T L Q K Q V S Y S L A K F N D F S I N G K L G S I C G Q L L I L T Y V Y G K E T P D G I K I T L D N L T M Q E L G Y S S G I A H S S A V S R I I S K L K Q E K V I V Y K N S C F Y V Q N L D Y L K R Y A P K L D E W F Y L A C P A T W G K L N. (SEQ ID NO: 33) 1 atgaacgctc aagcagaaga attcaaaaaa tatttagaaa ctaacgggat aaaaccaaaa 61 caatttcata aaaaagaact tatttttaac caatgggatc cacaagaata ttgtattttt 121 ctatatgatg gtatcacaaa gctcacgagt attagcgaga acgggaccat catgaattta 181 caatactaca aaggggcttt cgttataatg tctggcttta ttgatacaga aacatcggtt 241 ggctattata atttagaagt cattagcgag caggctaccg catacgttat caaaataaac 301 gaactaaaag aactactgag caaaaatctt acgcactttt tctatgtttt ccaaacccta 361 caaaaacaag tttcatacag cctagctaaa tttaatgttt tttcgattaa cgggaagctt 421 ggctctattt gcggtcaact tttaatcctg acctatgtgt atggtaaaga aactcctgat 481 ggcatcaaga ttacactgga taatttaaca atgcaggagt taggatattc aagtggcatc 541 gcacatagct cagctgttag cagaattatt tccaaattaa agcaagagaa agttatcgtg 601 tataaaaatt catgctttta tgtacaaaat cgtgattatc tcaaaagata tgcccctaaa 661 ttagatgaat ggttttattt agcatgtcct gctacttggg gaaaattaaa ttaa (SEQ ID NO: 2) MKKIMLVFITLILVSLPIAQQTEAKDASAFNKENSISSVAPPASPPASPK TPIEKKHADEIDKYIQGLDYNKNNVLVYHGDAVTNVPPRKGYKDGNEYIV VEKKKKSINQNNADIQVVNAISSLTYPGALVKANSELVENQPDVLPVKRD SLTLSIDLPGMTNQDNKIVVKNATKSNVNNAVNTLVERWNEKYAQAYSNV SAKIDYDDEMAYSESQLIAKFGTAFKAVNNSLNVNFGAISEGKMQEEVIS FKQIYYNVNVNEPTRPSRFFGKAVTKEQLQALGVNAENPPAYISSVAYGR QVYLKLSTNSHSTKVKAAFDAAVSGKSVSGDVELTNIIKNSSFKAVIYGG SAKDEVQIIDGNLGDLRDILKKGATFNRETPGVPIAYTTNFLKDNELAVI KNNSEYIETTSKAYTDGKINIDHSGGYVAQFNISWDEVNYD. (SEQ ID NO: 4) MKKIMLVFITLILVSLPIAQQTEAKDASAFNKENSISSVAPPASPPASPK TPIEKKHADEIDKYIQGLDYNKNNVLVYHGDAVTNVPPRKGYKDGNEYIV VEKKKKSINQNNADIQVVNAISSLTYPGALVKANSELVENQPDVLPVKRD SLTLSIDLPGMTNQDNKIVVKNATKSNVNNAVNTLVERWNEKYAQAYSNV SAKIDYDDEMAYSESQLIAKFGTAFKAVNNSLNVNFGAISEGKMQEEVIS FKQIYYNVNVNEPTRPSRFFGKAVTKEQLQALGVNAENPPAYISSVAYGR QVYLKLSTNSHSTKVKAAFDAAVSGKSVSGDVELTNIIKNSSFKAVIYGG SAKDEVQIIDGNLGDLRDILKKGATFNRETPGVPIAYTTNFLKDNELAVI KNNSEYIETTSKAYTD. EXPERIMENTAL DETAILS SECTION
Example 1
LLO-Antigen Fusions Induce Anti-Tumor Immunity
Materials and Experimental Methods
Examples 1-2
Cell Lines
Western Blotting
Measurement of Tumor Growth
Effects of
51Cr Release Assay
TC-1-Specific Proliferation
Flow Cytometric Analysis
B16F0-Ova Experiment
Statistics
Results
Example 2
Lm-LLO-E7 Treatment Elicits TC-1 Specific Splenocyte Proliferation
Example 3
Fusion of E7 to LLO, ActA, or a Pest Amino Acid Sequence Enhances E7-Specific Immunity and Generates Tumor-Infiltrating E7-Specific CD8+ Cells
Materials and Experimental Methods
Results
Example 4
Passaging of
Materials and Experimental Methods
Bacterial Culture
Passage of Bacteria in Mice
Intracellular Cytokine Stain for IFN-Gamma
Results
Passaging in Mice Increases the Virulence of Recombinant
Example 5
A PrfA-Containing Plasmid is Stable in an LM Strain with a PrfA Deletion in the Absence of Antibiotics
Materials and Experimental Methods
Bacteria
Replica Plating
Results
Materials and Methods
Examples 6-10
PCR Reagents
Primers ADV451, 452 and 453. Orien- Specifi- Primer tation Sequence (5′ → 3′) city ADV451 Forward CCTAGCTAAATTTAATGT D133V (SEQ ID NO: 28) mutation ADV452 Forward CCTAGCTAAATTTAATGA Wild-type (SEQ ID NO: 29) sequence ADV453 Reverse TAATTTTCCCCAAGTAGCAGG Shared (SEQ ID NO: 30) sequence PCR reagents. Catalog Description Provider number 1 0.2 ml thin-walled PCR tubes: Applied N801-0612 GeneAmp autoclaved reaction Biosystems tube with cap 2 Water (PCR reagent) Sigma W1754 3 Taq DNA Polymerase with 10x reaction Sigma D1806 buffer containing 15 mM MgCl2 4 Set of deoxynucleotides (dNTPs), Sigma D7295 10 mM each 5 Primers ADV451, ADV452 Invitrogen and ADV453 6 Template DNA, midipreparations of pGG55 plasmids 7 Thermal cycler PTC200 MJ Research (48 wells block) Plasmid DNA Preparation
Set of individual PCR reactions to validate the method to detect the presence of wild-type prfA sequence in Lm-LLO-E7 samples. Expected Tube Primer A Primer B Template DNA Function result 1 ADV451 ADV453 1 ng of pGG55 Positive control for Positive (D133V) the ADV451 reaction 2 ADV452 ADV453 1 ng of pGG55 Negative control for Negative (D133V) the ADV452 reaction (specificity) 3 ADV452 ADV453 1 ng of pGG55 Positive control for Positive (wild-type) + 1 ng the ADV452 reaction of pGG55 (D133V) 4 ADV452 ADV453 100 pg of pGG55 Test the sensitivity of Positive (wild-type) + 1 ng the reaction of pGG55 (D133V) 5 ADV452 ADV453 10 pg of pGG55 Test the sensitivity of Positive (wild-type) + 1 ng the reaction of pGG55 (D133V) 6 ADV452 ADV453 1 pg of pGG55 Test the sensitivity of Positive (wild-type) + 1 ng the reaction of pGG55 (D133V) 7 ADV452 ADV453 100 fg of pGG55 Test the sensitivity of Positive (wild-type) + 1 ng the reaction pGG55 (D133V) 8 ADV452 ADV453 10 fg of pGG55 Test the sensitivity of Positive (wild-type) + the reaction pGG55 (D133V) 9 ADV452 ADV453 1 fg of pGG55 Test the sensitivity of Weakly (wild-type) + the reaction positive pGG55 (D133V) 10 ADV452 ADV453 0.1 fg of pGG55 Test the sensitivity of To be (wild-type) + the reaction determined pGG55 (D133V) Master PCR mix preparation. Reagent Quantity (μl) Water 206.25 Taq DNA Polymerase 10x reaction buffer 27.5 containing 15 mM MgCl2 Deoxynucleotides (dNTPs) 10 mM each 5.5 Primers ADV452 (20 μM in water) 5.5 Primers ADV453 (20 μM in water) 5.5 pGG55 D133V (Lm-LLO-E7) plasmid (1 ng/μl) 11 Taq DNA Polymerase (5 U/μl) 2.75 Total 264 PCR protocol for validation of the method to detect the presence of wild-type prfA sequence using primers ADV451, 452 and 453. Reagent PCR Water 18.75 μl PCR Buffer 10x + MgCl215 mM 2.5 μl Deoxynucleotides mix (dATP, dCTP, dGTP and dTTP) 0.5 μl 10 mM each Primer ADV452 (20 μM) 0.5 μl Primer ADV453 (20 μM) 0.5 μl Taq DNA polymerase (5 U/μl) 0.25 μl Template DNA (1 ng/μl) pGG55 D133V 1 μl Template DNA pGG55 WT (tubes 3 to 10)a 1 μl Final volume per tubeb 25 μl apGG55 WT (1 ng in tube 3; 100 pg in tube 4; 10 pg in tube 5; 1 pg in tube 6; 100 fg in tube 7; 10 fg in tube 8; 1 fg in tube 9; 0.1 fg in tube 10). bIn tube 1, add 0.5 μl of water and 0.5 μl of primer ADV451 (20 μM stock); in tube 2 add 1 μl of water. PCR cycling conditions to detect the presence of wild- type prfA sequence using primers ADV451, 452 and 453. Step Temperature Time Number of cycles 1. 94° C. 2 minutes and 30 seconds 1 2. 94° C. 30 seconds 1 3. 53° C. 30 seconds 1 4. 72° C. 30 seconds 1 5. Repeat steps 2 to 4 12 6. 94° C. 30 seconds 1 7. 50° C. 30 seconds 1 8. 72° C. 30 seconds 1 9. Repeat steps 6 to 8 23 10. 72° C. 10 minutes 1 Sequencing:
Results
Example 6
Sequencing is not a Sensitive Method to Detect the Reversion of the D133V Mutation
Example 7
Development of a Highly Specific and Sensitive PCR Method to Detect Reversion of the D133V Mutation
Example 8
Specificity of the PCR Method
Example 9
Sensitivity of the PCR Method
Example 10
Recombinant