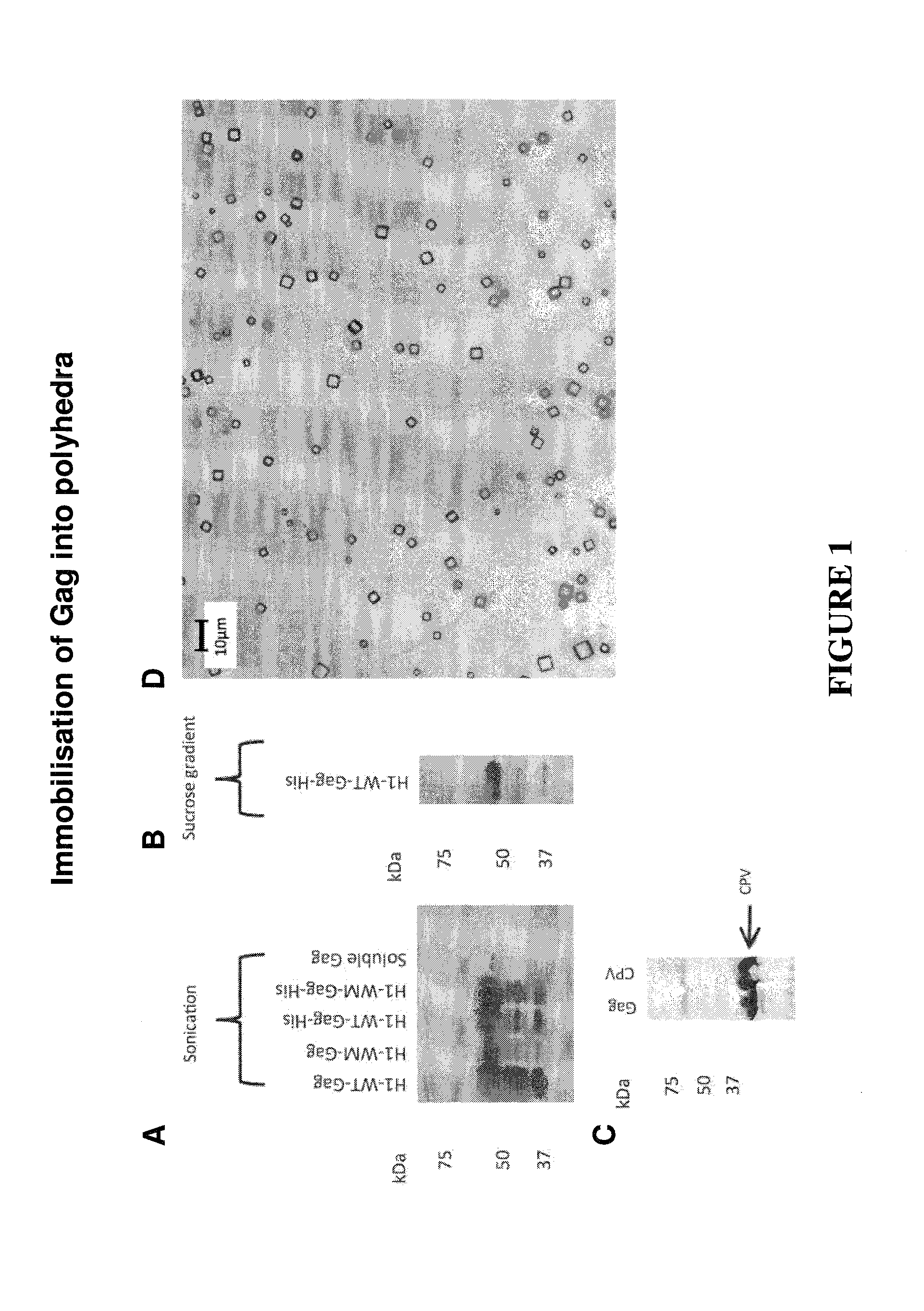
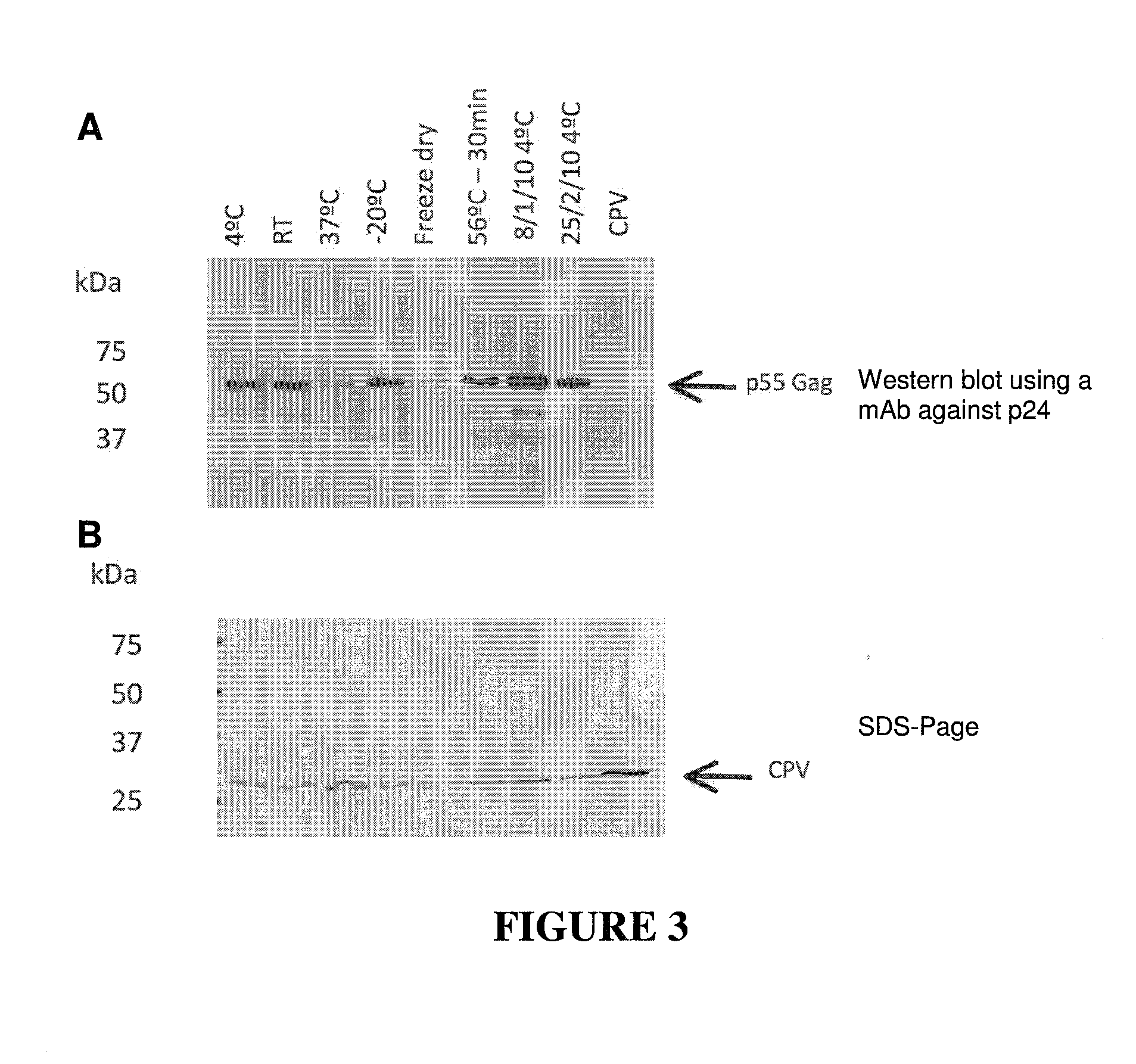
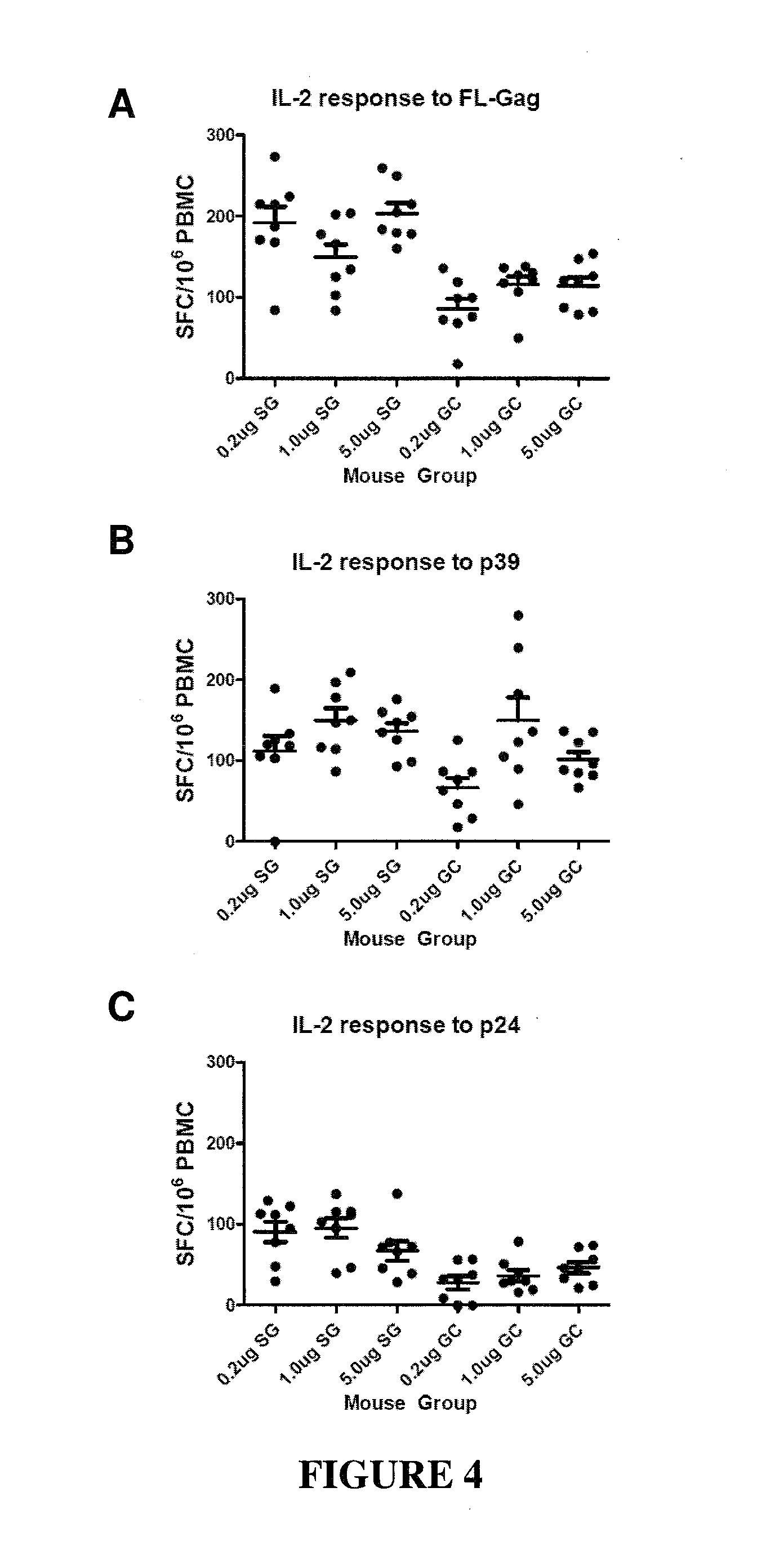
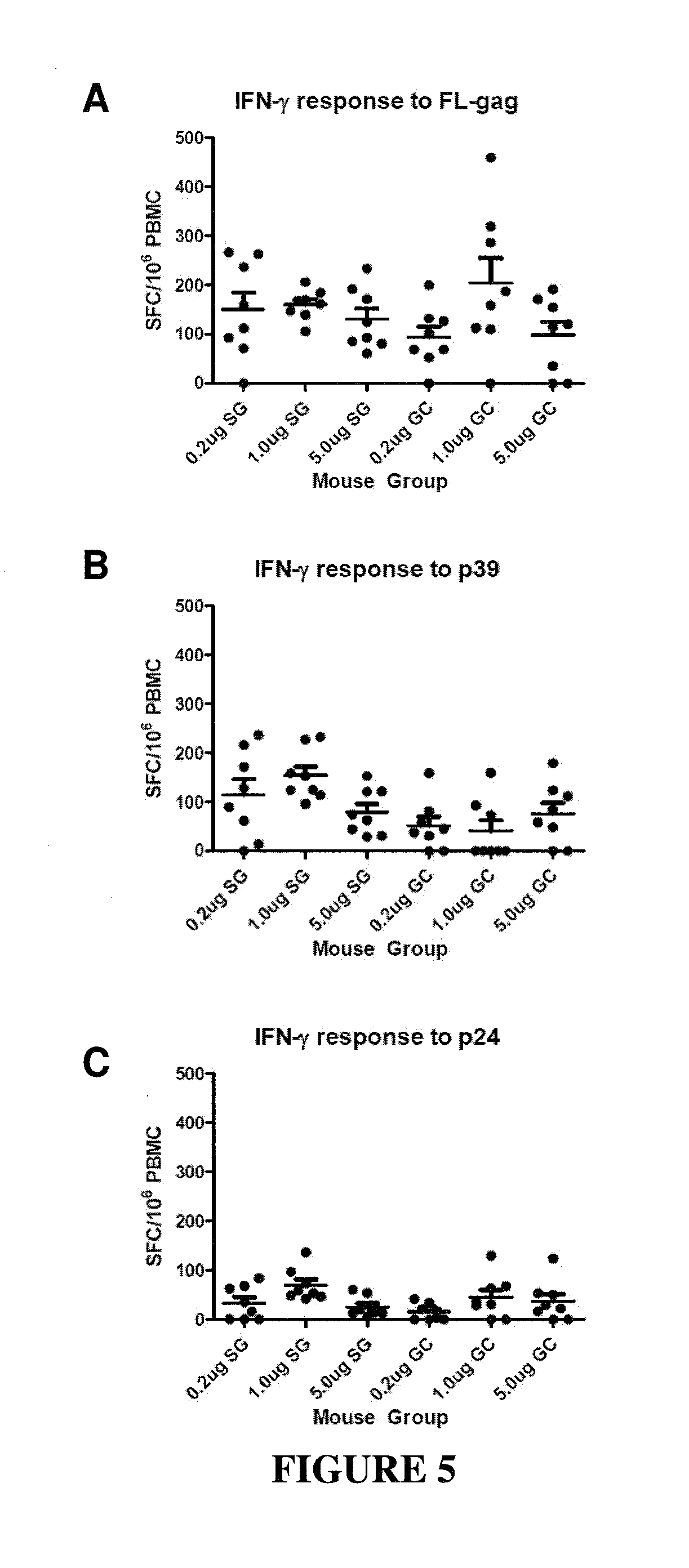
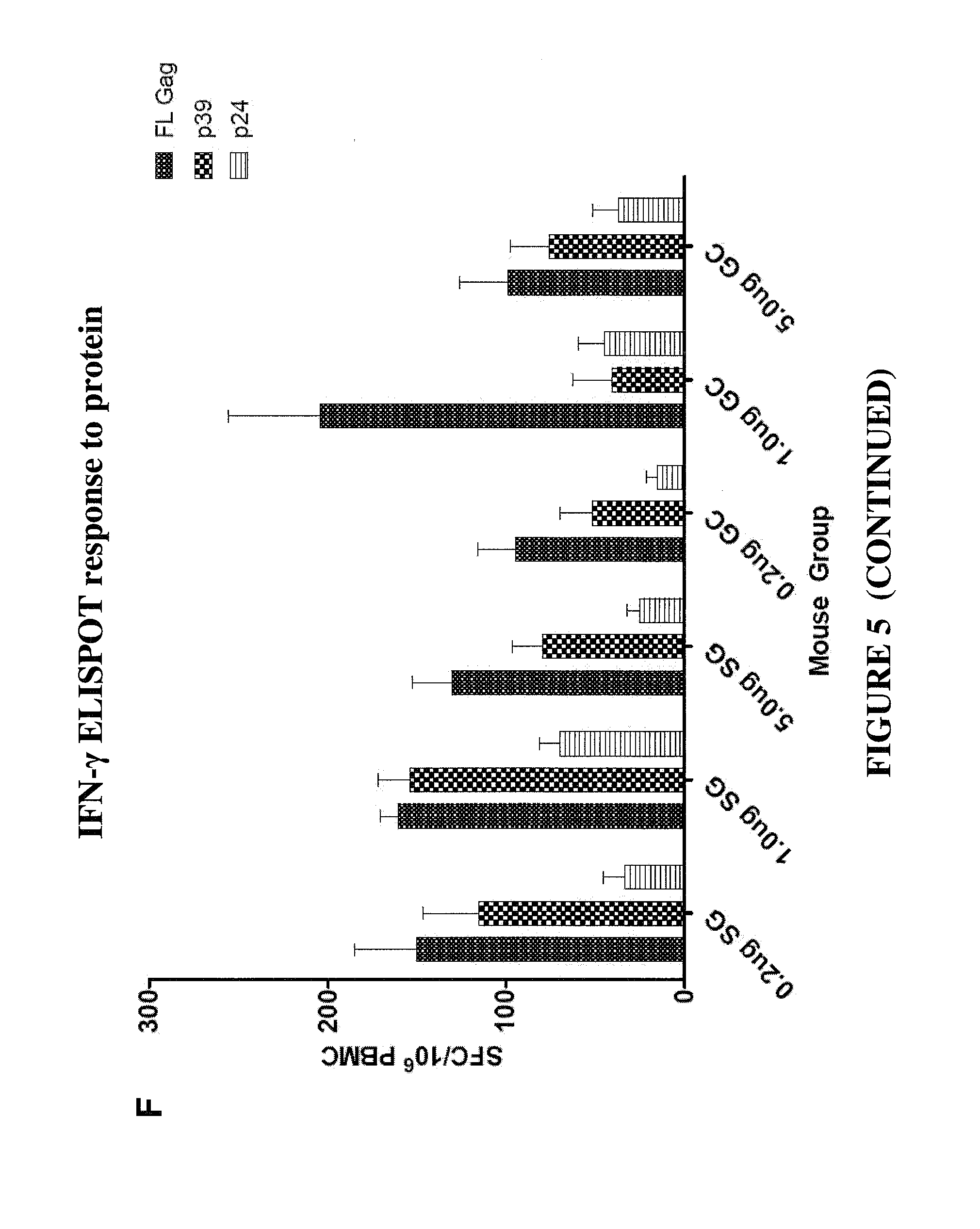
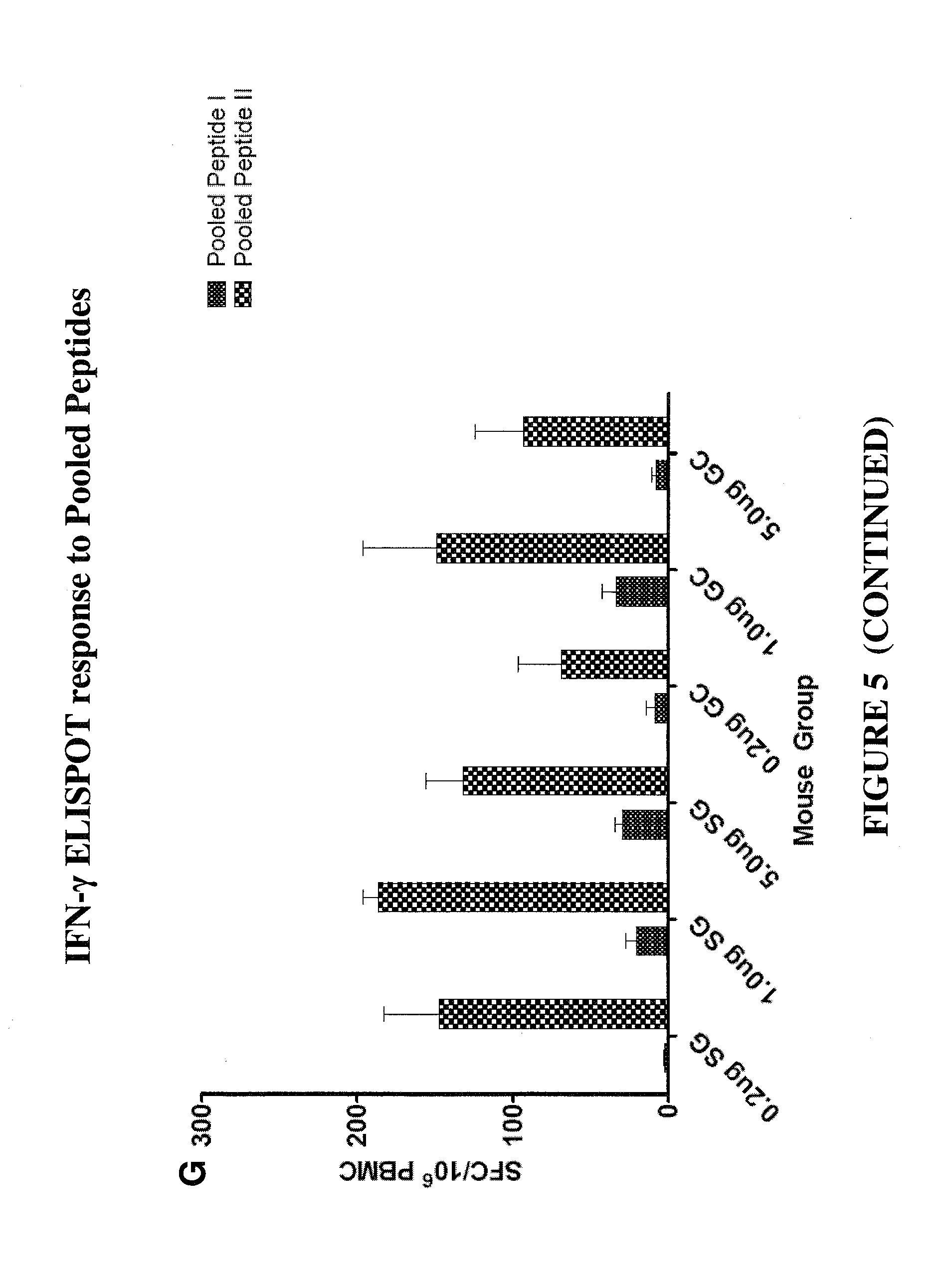
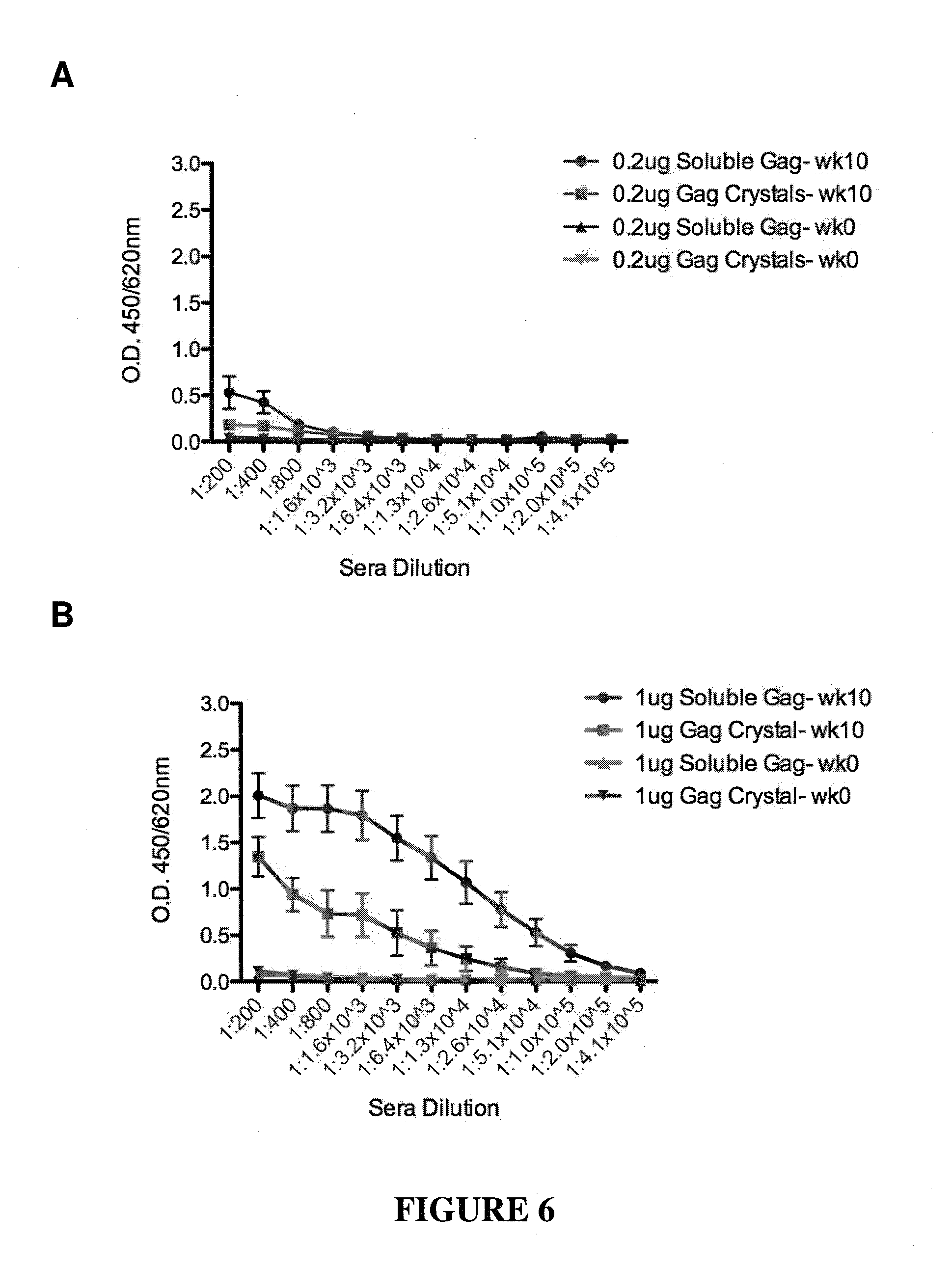
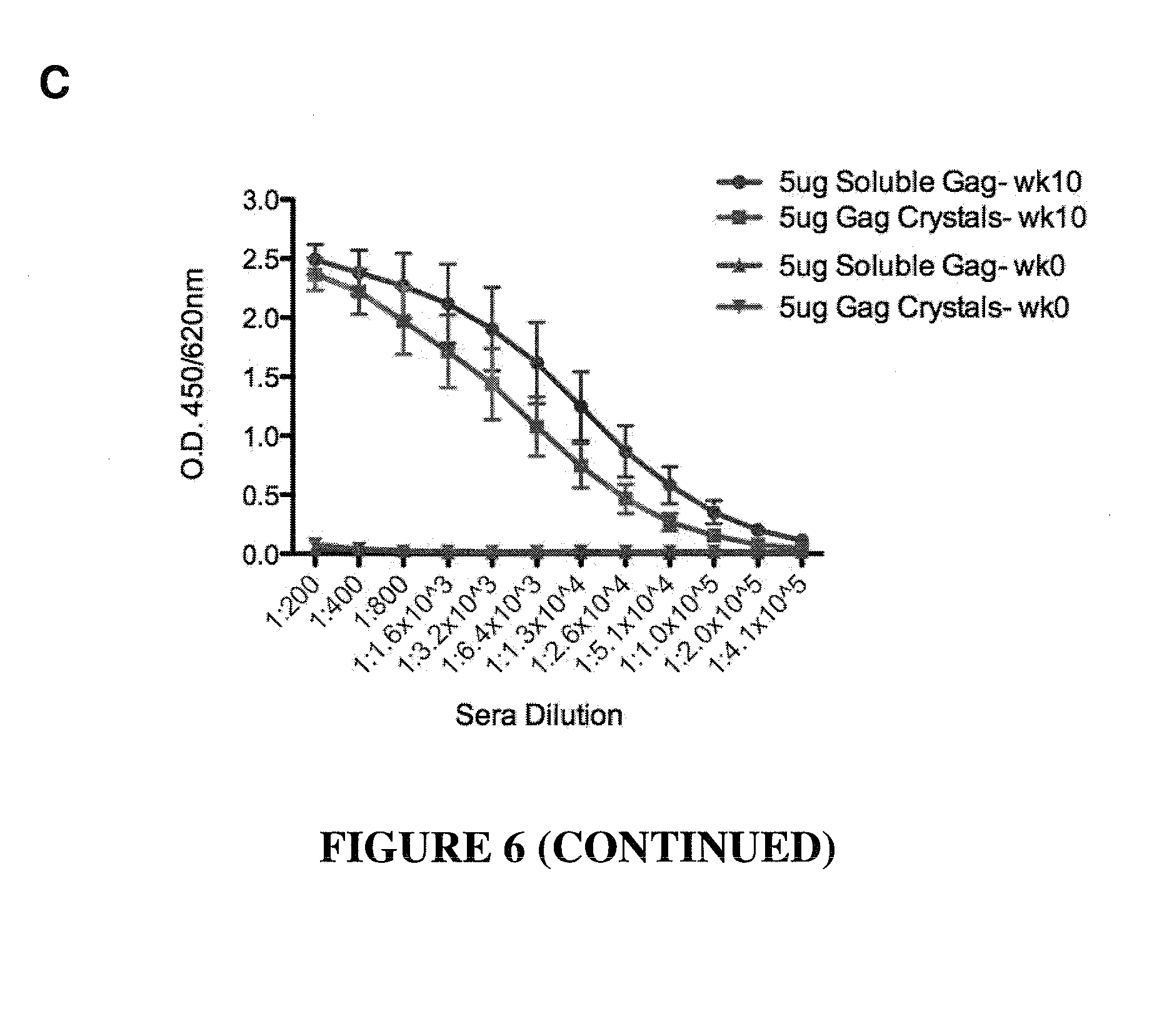
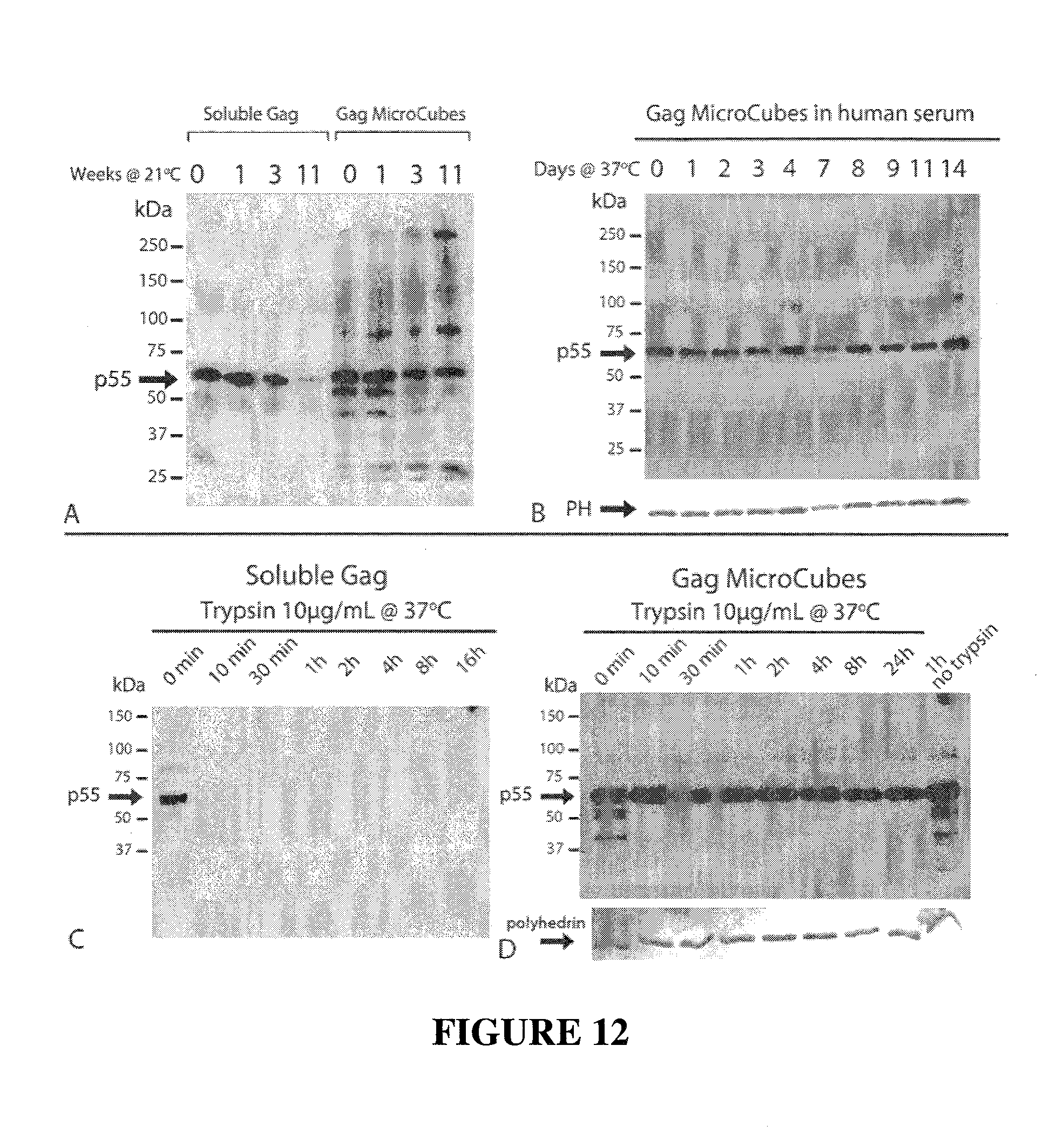
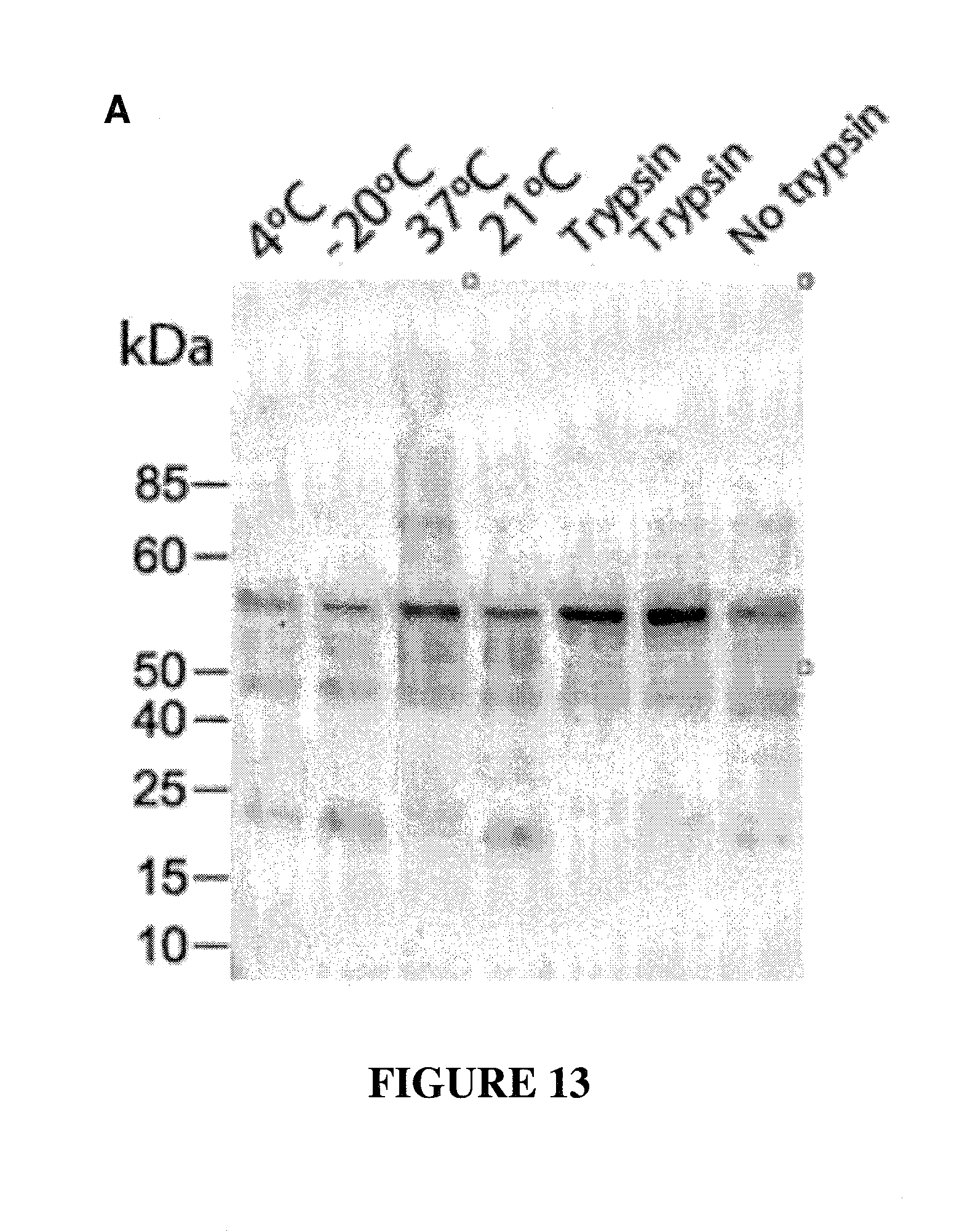
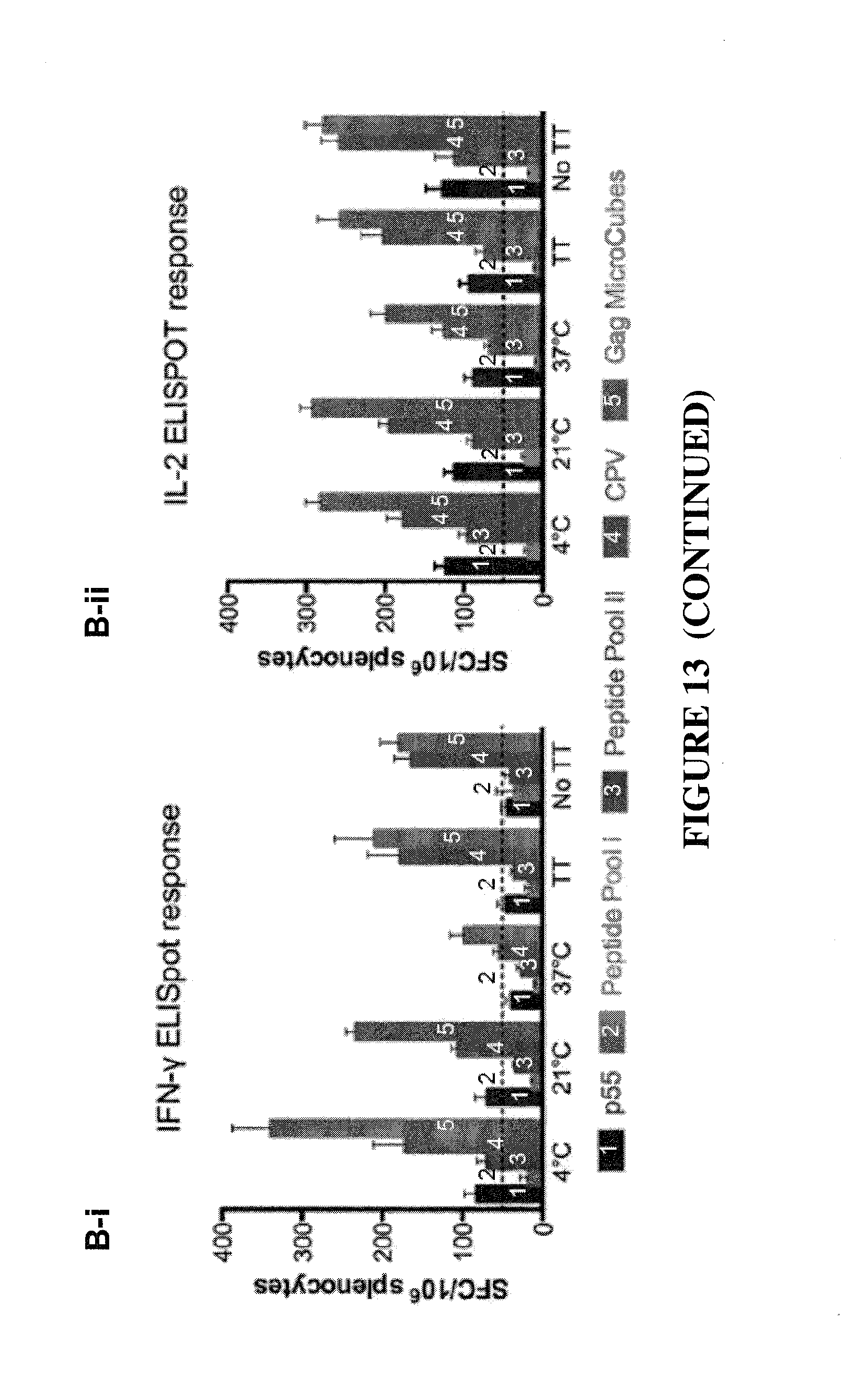
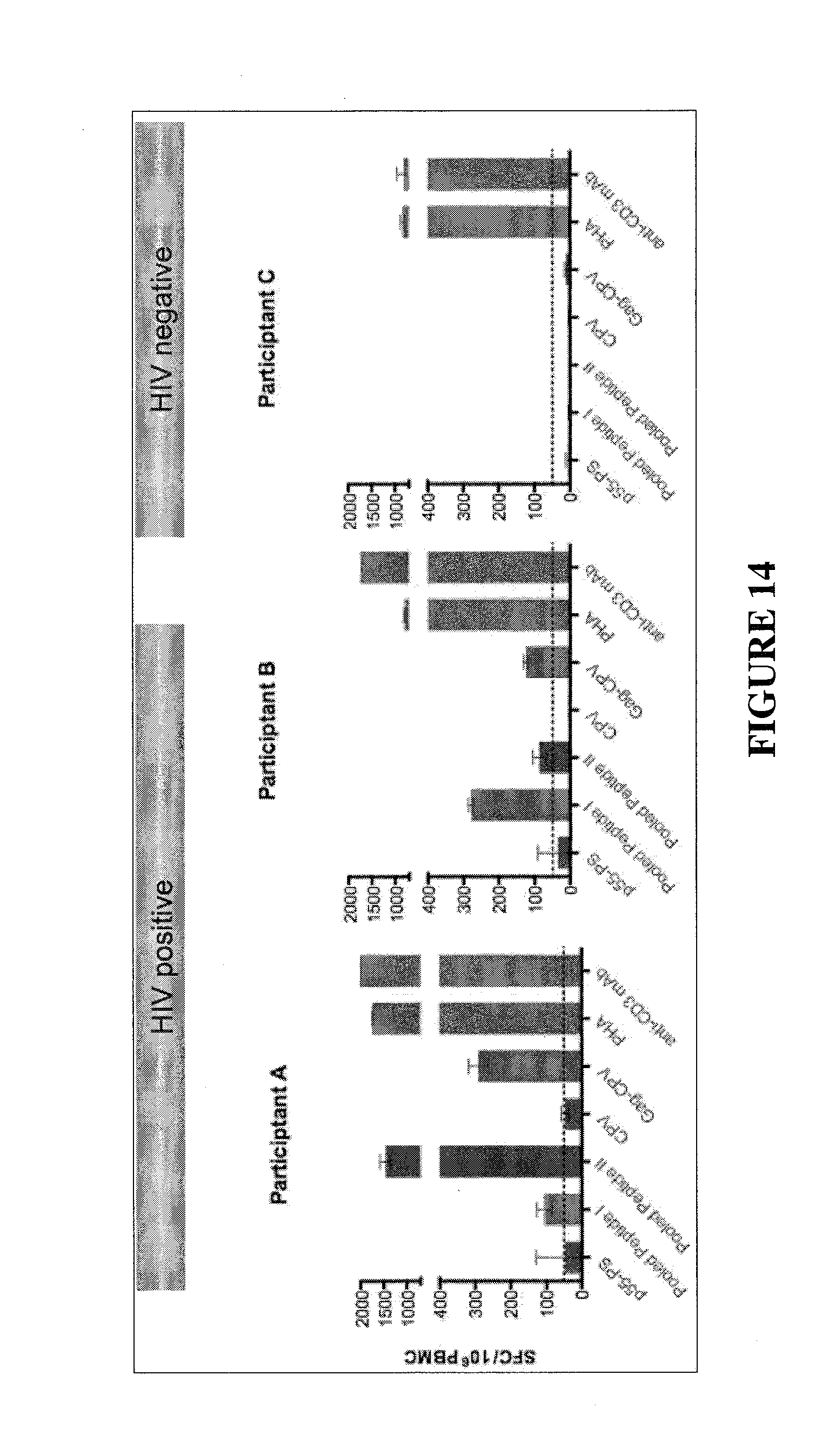
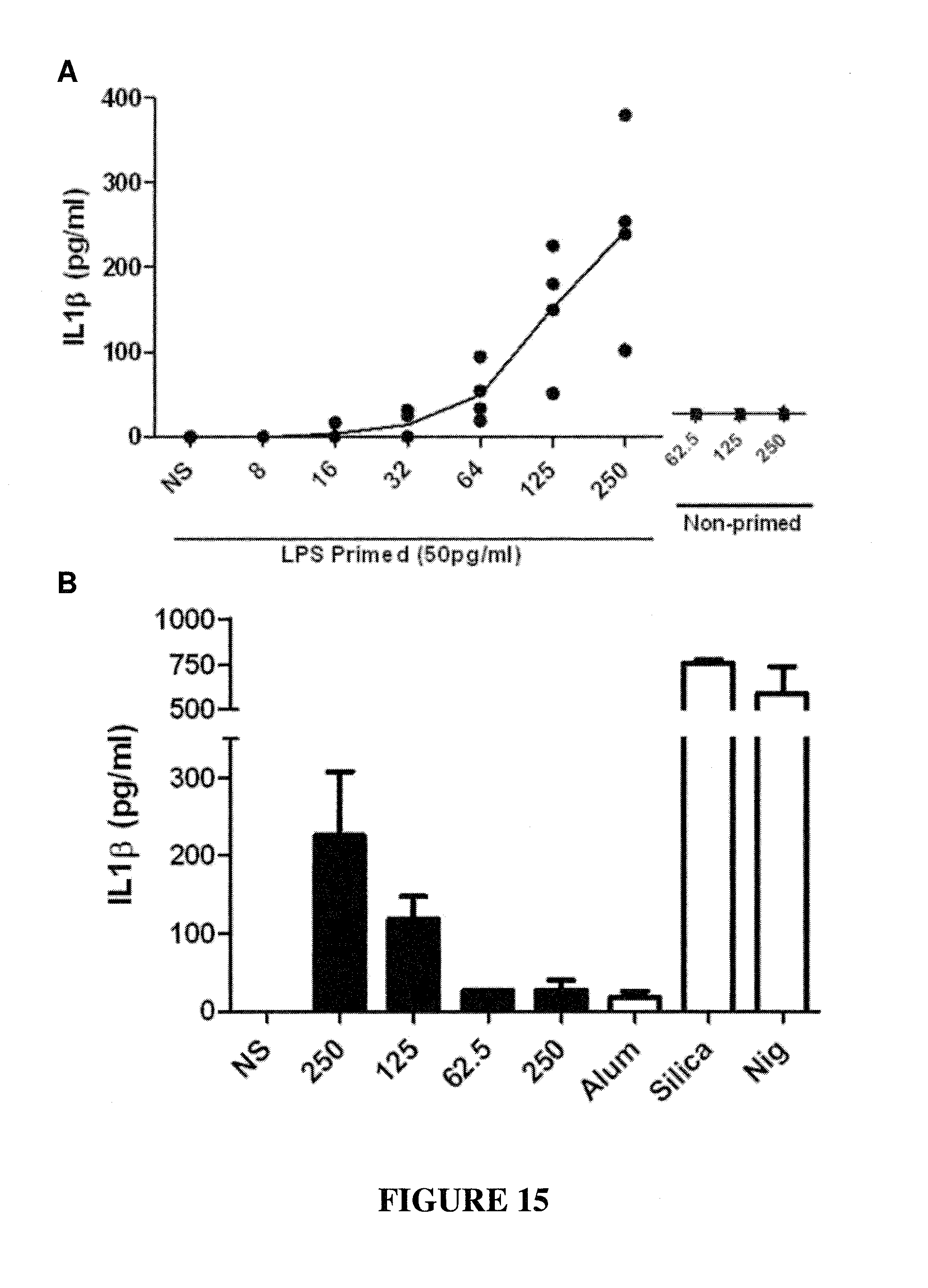
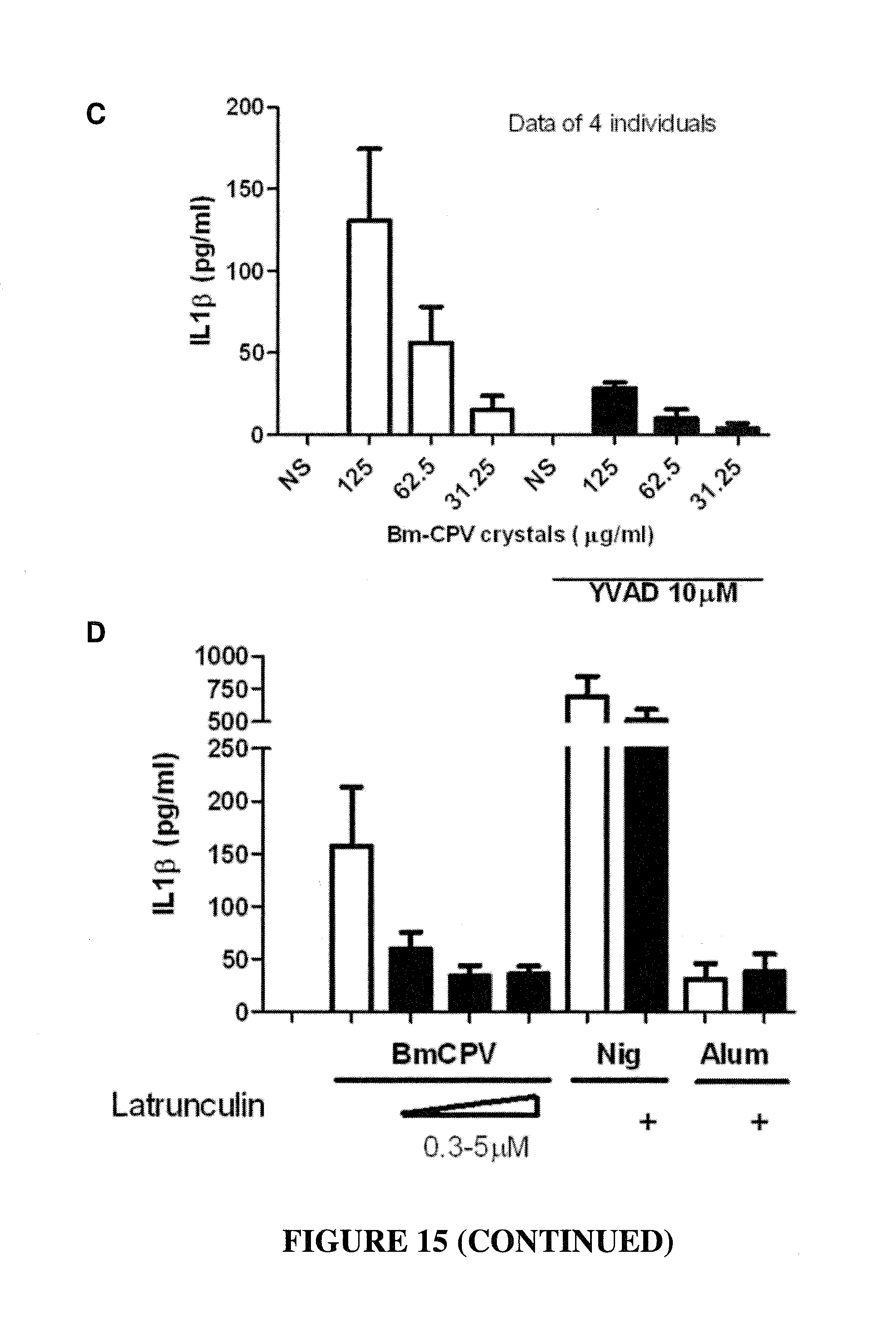
CONSTRAINED IMMUNOGENIC COMPOSITIONS AND USES THEREFOR
The present invention relates to immunogenic, proteinaceous, constrained complexes and to compositions and kits comprising them. In some embodiments, the invention relates to delivery of constrained antigens to subjects to induce an immune response. Bibliographic details of references in the subject specification are also listed at the end of the specification. The reference in this specification to any prior publication (or information derived from it), or to any matter which is known, is not, and should not be taken as an acknowledgment or admission or any form of suggestion that that prior publication (or information derived from it) or known matter forms part of the common general knowledge in the field of endeavour to which this specification relates. Despite much progress in understanding the mechanisms of immunity, vaccines against major pathogens such as HIV and There is a need for a versatile platform technology able to present antigens of various nature and size and induce robust humoral and cellular responses. Each embodiment in this specification is to be applied mutatis mutandis to every other embodiment unless expressly stated otherwise. The articles “a” and “an” are used herein to refer to one or to more than one (i.e. to at least one) of the grammatical object of the article. By way of example, “a cell” means one cell or more than one cell. An “antigen” means one antigen or more than one antigen. Throughout this specification, unless the context requires otherwise, the words “comprise,” “comprises” and “comprising” will be understood to imply the inclusion of a stated step or element or group of steps or elements but not the exclusion of any other step or element or group of steps or elements. Thus, use of the term “comprising” and the like indicates that the listed elements are required or mandatory, but that other elements are optional and may or may not be present. By “consisting of” is meant including, and limited to, whatever follows the phrase “consisting of”. Thus, the phrase “consisting of” indicates that the listed elements are required or mandatory, and that no other elements may be present. By “consisting essentially of” is meant including any elements listed after the phrase, and limited to other elements that do not interfere with or contribute to the activity or action specified in the disclosure for the listed elements. Thus, the phrase “consisting essentially of” indicates that the listed elements are required or mandatory, but that other elements are optional and may or may not be present depending upon whether or nut they affect the activity or action of the listed elements. The present invention relates broadly to the use of elements of insect virus crystals, referred to as polyhedra, to present antigens associated with pathogens, diseases or conditions affecting human or non-human mammalian subjects and to induce immune responses. The ability of antigen-polyhedrin complexes in the form of polyhedra (herein also referred to as MicroCubes) to elicit an immune response is surprising as it was assumed that the crystals would be rapidly cleared from the organism, or be toxic, or be unable to be processed by antigen presenting cells, or be capable of eliciting only either a humoral or a cellular immune response. In some embodiments, polyhedral polyhedrin and a polypeptide comprising an antigen form a stable complex which at least partially constrains the structure of the antigen and/or protects the antigen from degradation. Thus, in some embodiments, the present invention provides a vehicle for presenting antigens of interest to the immune system. In nature, viral polyhedra contain multiple viral particles embedded (occluded) within the crystalline lattice which acts as a survival and transmission mechanism. The encapsulated viral particles can remain infectious in soil for many years and the life cycle is continued when an insect ingests the crystals that break down in the alkaline mid-gut to release infective viral particles. As known in the art, polyhedrin targeting peptides (tags) can be used to draw fusion proteins comprising them into a crystal structure comprising polyhedrin. The term “complex” refers to the “antigen-polyhedrin subunit” which forms the modified CPV polyhedrin as well as the “modified polyhedra”. In some preferred embodiments, the term “complex” refers to the modified polyhedra (the terms “MicroCubes”, “polyhedra crystals”, “modified polyhedra crystals”, “polyhedra”, “polyhedrin” or “micromolecular structure” and the like are used interchangeably) comprising the antigen of a pathogen or disease or condition affecting a human or non-human mammalian subject. In some embodiments, the present invention employs protocols developed previously to express polypeptides as fusion proteins in insect polyhedra. This technology is known in the art and may be reviewed for example in Ikeda et al. The present invention provides an immunogenic or vaccine composition comprising a complex comprising an antigen of a pathogen or other antigen against which a immune response is sought in a human or non-human animal subject and a polyhedrin or polyhedrin-like protein. “Polyhedrin” and “polyhedrin-like” encompasses any naturally occurring form of polyhedrin from any cytoplasmic polyhedrosis virus (cypo) (CPV) as well as their biologically active portions and variants, analogs, homologs or derivatives of these, as defined herein. Different polyhedrin polypeptide and peptide sequences are available in the art (see NCBI Entrez Search). A polyhedrin may be selected from the art and routinely tested in the methods described herein. Polyhedrin molecules produced by CPV are distinct from those produced by baculoviruses. They differ in structure and the viruses are unrelated. Differences are described between their molecular structures in Coulibaly et al., In some embodiments, the MicroCubes are in isolated, homogeneous, fully or partly purified form. Isolation and/or purification can be carried out by methods known in the art including salt fractionation, ion exchange chromatography, gel filtration, size-exclusion chromatography, size-fractionation, and affinity and immunoaffinity chromatography. FACS separation may also be employed. The term “isolated” or “purified” means material that is substantially or essentially free from components that normally accompany it in its native state. For example, an “isolated complex”, as used herein refers to a complex isolated from the cellular, cell-free, or molecular mixtures used in its production. In some embodiments, the purified complex is at least 95 to 99% pure. As noted, in one preferred embodiment, the polyhedrin is derived from a cytoplasmic polyhedrosis (cypo) virus (CPV). In another embodiment, the polyhedrin is not derived from a baculovirus. In an illustrative embodiment, delivery of the complex to a subject in substantially particulate polyhedral form induces an immune response thereto. In accordance with the present invention, the polyhedron reduces degradation of antigens. In some embodiments, it also activates the immune response and therefore potentially enhances the antigen-specific immune response. In an illustrative example, an antigen against which an immune response is sought is an antigen associated with a condition such as a tumor i.e., a tumor antigen. Accordingly, in some embodiments, the invention employs one or more antigens that are described in the art as candidate antigens for vaccination purposes because, for example, they engender an effective immune response in an animal model, and re-package the antigen(s) as a complex with polyhedrin that forms macromolecular polyhedra wherein the antigen is structurally and physically constrained. Without being bound by any particular theory or mode of action, it is proposed that delivery of antigen in particulate polyhedral form will induce enhanced cellular and/or immune responses, preferably both. Alternatively, or in addition, slow or sustained release of antigen from the micromolecular structure is proposed to reduce the need for multiple administrations and/or generate higher titre/strength cellular or antibody responses. In one embodiment the invention provides a stable immunogenic or vaccine composition comprising a complex comprising an antigen of a pathogen or other antigen against which a immune response is sought in a human or non-human animal subject and a polyhedrin protein derived from a cytoplasmic polyhedrosis virus (CPV) wherein delivery of the complex to a subject in substantially particulate polyhedral form induces an immune response thereto. Reference to “stable” includes that the antigen component of the complex in the polyhedron is substantially resistant to degradation under physiological or environmental conditions or exhibits decreased degradation compared to a control such as the antigen in the absence of the complex or polyhedra comprising same. In some embodiments, the antigen in the polyhedra is heat stable. For example, as described in the Examples, MicroCube antigens are stable at between about 4° C. and about 21° C. and even at about 37° C. In some other embodiments, the antigen in the polyhedra displays decreased degradation. In some embodiments, reference to “decreased degradation” refers to a composition displaying less than 50%, or less than 40%, less than 30%, less than 20%, less than 10%, less than 1% antigen degradation over a storage period under conditions wherein the same antigen not present in a complex with polyhedrin or in a polyhedron exhibits more than 50%, 60%, 70% or more antigen degradation. In some embodiments, the antigen in the polyhedron is resistant to enzymatic such as trypsin degradation. In an illustrative non-limiting embodiment, the polyhedrin is derived from By “derived from” is meant naturally occurring forms and functional variants of naturally occurring forms and therefore includes sequences directly or indirectly derived from an organism. For example, a viral polypeptide such as polyhedrin is “derived from” a particular polypeptide of a virus (viral polypeptide) if it is (i) encoded by an open reading frame of a polynucleotide of that virus (viral polynucleotide), or (ii) displays sequence and or structure-functional similarity to polypeptides of that virus as described herein. Functional variants are described herein and include derivatives which may be fragments of a polyhedrin polypeptide. In some embodiments, the antigen is fused to a polyhedrin targeting peptide such as the targeting peptide is derived from the N-terminal H1 α-helix or VP3 polyhedrin recognition signal of polyhedrin of CPV or is a functional variant thereof. In some embodiments, the targeting peptide is derived from the N-terminal H1 α-helix or VP3 polyhedrin recognition signal of polyhedrin of For the avoidance of doubt in some embodiments antigen-polyhedrin targeting fusion proteins are chimeric polypeptides by which is meant that the combination does not occur in nature and that the protein comprises an antigen from one organism and polyhedrin targeting peptide derived from a second organism, such as different species. In an illustrative embodiment, a chimeric antigen-polyhedrin targeting protein of the present invention is produced wherein at least two polypeptides or peptides derived from different species are linked by covalent bonds, either by being expressed as part of the same expression product or by synthesis. In both cases the resulting polypeptide may be referred to as a fusion protein. Direct attachment of antigen to polyhedra by covalent cross-linking or coating is also contemplated. The terms “polypeptide” “protein” and “peptide” and “glycoprotein” are used interchangeably and mean a polymer of amino acids not limited to any particular length. The term does not exclude modifications such as myristylatlon, glycosylation, phosphorylation and addition and/or deletion of signal sequences. A “part” or “portion” or “region” or “domain” of a polypeptide such as a polyhedrin H1 α-helix (tag) of cypovirus is defined as having a minimal size of at least about 10 amino acids or about 20 to 30 amino acids, 15 to 100 amino acids or about 5 to 80 amino acids or about 15 to 120 amino acids. As used herein, an “immune response” refers to the reaction of the body as a whole to the presence of a composition of the present invention which includes making antibodies and developing immunity to the composition. Immunity may develop as a result of activation of both innate and adaptive arms of the immune response. Therefore, an immune response to an antigen of a pathogen of a human or non-human animal or an antigen associated with a human condition (such as cancer) or disease as described herein also includes the development in a subject of a humoral and/or cellular immune response to the antigen of interest. Any such response can be modified, including enhanced or activated, by stimulation of an innate immune response. A “humoral immune response” is mediated by antibodies produced by plasma cells. A “cellular immune response” is one mediated by T lymphocytes and/or other white blood cells. An “immunological response” or “immune response” to an antigen includes the development in a subject of a humoral (B-cell) and/or a cellular immune (T-cell) response to an antigen. The polyhedral component of the subject complex is not an antigen as defined herein however as determined herein it elicits an immune response including cytokine secretion and or the activation of inflammasome potentially enhancing an antigen specific immune response as sought. The ability of a particular antigen to stimulate a cell-mediated or humoral immunological response, including the production of antibodies by plasma cells and B-cells, the activation of suppressor T-cells and/or γδT cells may be determined by any number of assays, such as by lymphoproliferation (lymphocyte activation) assays, CTL cytotoxic cell assays, or by assaying for T-lymphocytes specific for the antigen in a sensitized subject. Such assays are well known in the art. Methods of measuring cell-mediated immune response include measurement of intracellular cytokines or cytokine secretion by T-cell populations, or by measurement of epitope specific T-cells. The immune response may serve to neutralise infectivity, reduce transmission or load, and/or mediate antibody-complement, or antibody dependent cell cytotoxicity (ADCC) to provide protection to an immunised host. Immune responses can be determined using standard immunoassays and neutralisation assays, as known in the art. An “immunogenic composition” is a composition that comprises an antigenic molecule where administration of the composition to a subject results in the development in the subject of a humoral and/or a cellular immune response to the antigenic molecule of interest. In accordance with the present invention, the polyhedrin protein or peptide is also immunogenic and stimulates an immune response suitable for enhancing the immune response to the antigen against which an immune response is sought. Assays for assessing an immune response are described in the Examples and may comprise in vivo assays, such as assays to measure antibody responses and delayed type hypersensitivity responses. In an embodiment, the assay to measure antibody responses primarily may measure B-cell function as well as B-cell/T-cell interactions. For the antibody response assay, antibody titers in the blood may be compared following an antigenic challenge. As used herein, “antibody titers” can be defined as the highest dilution in post-immune sera that resulted in a value greater than that of pre-immune samples for each subject. These levels can be quantitated according to the type of antibody, as for example, IgG. IgG1, IgG2, IgG3, IgG4, IgM, IgA or IgD. Also, the development of immune systems may be assessed by determining levels of antibodies and lymphocytes in the blood without antigenic stimulation. The in vitro assays may comprise determining the ability of cells to divide, or to provide help for other cells to divide, or to release lymphokines and other factors, express markers of activation, and lyse target cells. Lymphocytes in mice and man can be compared in in vitro assays. In an embodiment, the lymphocytes from similar sources such as peripheral blood cells, splenocytes, or lymph node cells, are compared. It is possible, however, to compare lymphocytes from different sources as in the non-limiting example of peripheral blood cells in humans and splenocytes in mice. For the in vitro assay, cells may be purified (e.g., B-cells, T-cells, and macrophages) or left in their natural state (e.g., splenocytes or lymph node cells). Purification may be by any method that gives the desired results. The cells can be tested in vitro for their ability to proliferate using mitogens or specific antigens. The ability of cells to divide in the presence of specific antigens can be determined using a mixed lymphocyte reaction (MLR) assay. Supernatant from the cultured cells can be tested to quantitate the ability of the cells to secrete specific lymphokines. The cells can be removed from culture and tested for their ability to express activation antigens. This can be done by any method that is suitable as in the non-limiting example of using antibodies or ligands which bind to the activation antigen as well as probes that bind the RNA coding for the activation antigen. In some embodiments, phenotypic cell assays can be performed to determine the frequency of certain cell types. Peripheral blood cell counts may be performed to determine the number of lymphocytes or macrophages in the blood. Antibodies can be used to screen peripheral blood lymphocytes to determine the percent of cells expressing a certain antigen as in the non-limiting example of determining CD4 cell counts and CD4/CD8 ratios. Accordingly, the present invention provides a composition comprising a complex as herein described wherein the immune response to the complex includes a cellular and a humoral response. In some embodiments, the immune response to the polyhedrin or polyhedrin peptide portion of the complex comprises a cellular or humoral response. In some embodiments, the immune response to the polyhedrin or polyhedrin peptide portion of the complex comprises inflammasome activation. In some further embodiments, the composition comprising a pharmaceutically or physiologically acceptable carrier and/or diluent. The term “vaccine” as used herein refers to a pharmaceutical composition comprising an immunologically active component that induces an immunological response in a subject and possibly but not necessarily one or more additional components that enhance the immunological activity of said active component (for example an adjuvant). A vaccine may additionally comprise further components typical to pharmaceutical compositions. The immunologically active component of a vaccine according to the present invention comprises an antigen of a pathogen or other antigen against which a immune response is sought in a human or non-human animal subject and a polyhedrin protein. The terms “vaccine” and “vaccine composition” are used interchangeably in the present invention. As determined herein, the polyhedrin portion also induces an immune response “Subjects” contemplated in the present invention include any animal of commercial or humanitarian interest including conveniently, primates, livestock animals including fish, crustacea, and birds, laboratory test animals, companion animals, or captive wild animals. In some embodiments the subject is a mammalian animal. In particular embodiments, the subject is a human subject. In some embodiments, “subjects” are humans or animals including laboratory or art accepted test or vehicle animals. “Patients” include human subjects in need of treatment or prophylaxis. In another embodiment, the invention provides an immunogenic composition comprising an antigen of a pathogen or other antigen against which an immune response is sought and a CPV polyhedron wherein delivery of the composition induces an immune response to the antigen and wherein the CPV polyhedron enhances the immune response to the antigen. In some embodiments, the invention provides an Immunogenic composition comprising CPV polyhedra for use in conjunction with an antigen to stimulate an immune response to the antigen. In some embodiments, the CPV polyhedron is derived from In another embodiment, the present invention provides an immunogenic composition as described herein comprising a complex comprising an antigen of a pathogen or other antigen against which a immune response is sought in a human or non-human animal subject and a polyhedrin protein for use in the manufacture of a vaccine for the treatment or prevention of an infection, disease or condition associated with the antigen. In another embodiment, there is provided for a use of an immunogenic composition as described herein comprising a complex comprising an antigen of a pathogen or other antigen against which a immune response is sought in a human or non-human animal subject and a polyhedrin protein in the manufacture of a medicament for the treatment or prevention of an infection, disease or condition associated with the antigen. In another broad embodiment, there is provides a method for eliciting an immune response in a subject or patient, the method comprising administering to the subject or patient an effective amount of an immunogenic composition as described herein comprising a complex comprising an antigen of a pathogen or other antigen against which a immune response is sought in a human or non-human animal subject and a polyhedrin protein, under conditions to elicit an immune response. Further, the invention includes method for immunising a subject against infection or disease or condition associated with the antigen comprising administering to the subject an immunogenic composition as described herein comprising a complex comprising an antigen of a pathogen or other antigen against which a immune response is sought in a human or non-human animal subject and a polyhedrin protein. Furthermore, the present invention provides a method for treating or preventing infection by a pathogen or a disease (cancer) or other condition comprising administering to the subject an immunogenic composition as described herein comprising a complex comprising an antigen of a pathogen or other antigen against which a immune response is sought in a human or non-human animal subject and a polyhedrin protein, for a time and under conditions sufficient to treat or prevent the infection or cancer/disease or condition. In one embodiment, the present invention provides a method for inducing an immune response in a subject, the method comprising administering to the subject an effective amount of a complex comprising (a) an antigen of a pathogen or other antigen associated with a condition against which an immune response is sought; and (b) polyhedrin, wherein administration is for a time and under conditions sufficient for the antigen to induce an immune response. In another embodiment, the present invention provides a method for inducing an immune response in a subject, the method comprising administering to the subject an effective amount of a complex comprising (a) a chimeric fusion polypeptide comprising a polyhedrin targeting peptide and an antigen of a pathogen or other antigen associated with a condition against which an immune response is sought; and (b) polyhedrin, wherein administration is for a time and under conditions sufficient for the antigen to induce an immune response. In a similar embodiment, the invention provides a method for treatment or prophylaxis of a viral infection in a subject comprising administering a complex comprising a virus antigen and/or fusion protein comprising same according to the present invention for a time and under conditions sufficient to treat or prevent the virus infection. In a similar embodiment, the invention provides a method for treatment or prophylaxis of an infection in a subject comprising administering a complex comprising a pathogen antigen or a tumor/cancer antigen and/or fusion protein comprising same according to the present invention for a time and under conditions sufficient to treat or prevent the pathogen or tumor/cancer infection/condition. In other similar embodiments, the invention provides the subject complexes and fusion proteins for use in the treatment and/or prophylaxis of a viral infection or a pathogen or tumor/cancer infection/condition. In further similar embodiments, the complexes and/or fusion proteins are proposed for use in the manufacture of a medicament for treatment and/or prophylaxis of a viral pathogen or other pathogenic infection or tumor. In some embodiments, the invention provides pharmaceutical compositions including immunogenic or putative vaccine compositions comprising an isolated nucleic acid molecule encoding the subject fusion polypeptide. In some embodiments, pharmaceutical compositions including an immunogenic or putative vaccine composition are formulated with a pharmaceutically acceptable carrier and/or diluent. In other embodiments, the present invention provides a pharmaceutical composition comprising a subject complex or fusion polypeptide as described herein. A putative vaccine composition is one, for example, that shows promise of inducing an effective immune response in an accepted animal or cellular model. In other embodiments, the invention provides a method for producing an isolated or purified antibody or immune cell that specifically binds to an antigen of a pathogen or other antigen against which an immune response is sought in a human or non-human animal subject or patient. The method comprises administering to a subject an effective amount of an immunogenic composition as described herein comprising a complex comprising an antigen of a pathogen or other antigen against which a immune response is sought in a human or non-human animal subject and a polyhedrin protein, and isolating or purifying antibody or immune cells. In some embodiments, the complex or polyhedra comprising same is in isolated, homogeneous, fully or partly purified form. In preferred embodiments, the polyhedrin is derived from a CPV. In further embodiments, delivery of the complex to a subject in substantially polyhedral form induces an immune response to the complex. In some embodiments, the antigen is fused to a polyhedrin targeting peptide. In some embodiments, the immune response to the complex includes an immune response to the polyhedrin portion of the complex and comprises a cellular or humoral immune response and/or comprises inflammasome activation. Activation may be detected by various assays such as by assaying for IL-1β secretion. In some embodiments, the polyhedrin is not targeted to the nucleus of insect cells and does not form a polyhedral envelope. In some embodiments, the immune response is a humoral and a cellular immune response. The above method encompasses the production of antibodies and/or immune cells in a non-human subject. In this embodiment, antibodies, for example, are suitable for use in the manufacture of therapeutic or prophylactic antibodies. In some other embodiments, such antibodies are useful for diagnosis, screening and research. In yet another embodiment, the methods encompass the induction of a humoral and/or immune response to the antigen in a subject susceptible to the pathogen or condition or in need of treatment or prophylaxis. In the case of prophylactic or therapeutic administration, mammalian including human subjects are particularly contemplated. In another embodiment, the present invention provides a fusion polypeptide comprising (a) a viral polyhedrin targeting peptide and (b) an antigen of a pathogen or other molecule against which an immune response is sought. In some embodiments, the fusion polypeptide is provided in a composition suitable for administration to a subject to inducing an immune response in the subject. Illustrative compositions comprise an adjuvant suitable for animal or human application as known in the art. Other illustrative compositions are formulated for delivering to mucosa such as of the nose, mouth, gut, etc. In an illustrative embodiment, trimeric polyhedrin polypeptides are organised around a scaffold of an N-terminal helix. Polyhedra are micromolecular complexes. Trimers are organised into tetrahedral clusters of tour trimers cross-linked by intermolecular disulphide bonds (Coulibaly et al, In another embodiment, the present invention provides a complex comprising (a) a fusion polypeptide comprising a polyhedrin targeting peptide and an antigen of a pathogen or other antigen associated with a condition against which an immune response is sought; and (b) polyhedrin. In some embodiments, the complex is immunogenic and/or provides sustained release in a subject. In other embodiments, the complex is suitable for eliciting an enhanced immune response compared to the immune response produced by the antigen not in the form of a complex with polyhedrin nor in the form of a fusion protein with a polyhedrin targeting peptide. In some embodiments, the complex is in the form of a recombinant or modified polyhedron comprising a plurality of fusion polypeptides comprising an antigenic portion and a polyhedrin targeting portion. In some embodiments, the antigen portion comprises one or more epitopes derived from a single pathogenic organism or condition. In other embodiments, the antigen portion comprises one or more epitopes from more than one pathogen or condition. In some embodiments, the recombinant or modified polyhedra in the size range of 0.1 um to 50 um, more particularly, 0.1 um to 10 um, depending upon the insect polyhedrin molecules employed. Particle size may be tailored to the mode of administration for immunisation. In an illustrative embodiment, the pathogen is HIV. In a further illustrative embodiment, the antigen is HIV Gag polypeptide or an antigenic peptide thereof. As known in the art a Gag is produced as a precursor comprising a myristylated protein (p55), which is typically processed to varying degrees by proteases to form matrix protein (MA-p17), core antigen capsid protein (CA-p24), nucleo-capsid protein (NC-p7), p6, p2 and p1. HIV Gag p39 comprises p24, p9 and p6. In another embodiment, the invention provides a method for producing a complex comprising (a) a fusion polypeptide comprising a polyhedrin targeting peptide and an antigen of a pathogen or other antigen associated with a condition against which an immune response is sought; and (b) polyhedrin, the method comprising expressing a nucleic acid molecule encoding the antigen as a fusion polypeptide with a polyhedrin targeting peptide and expressing a nucleic acid molecule encoding a polyhedrin or polyhedrin-like polypeptide in an insect or other suitable host cell and contacting the polyhedrin and fusion polypeptides for a time and under conditions sufficient for the fusion protein comprising the antigen and the polyhedrin to form a complex. In some embodiments, the two proteins are co-produced in an insect or other equivalent host cell. The complex typically comprises a plurality of copies of the fusion protein. In particular embodiments, the method further comprised isolating or purifying the complex from other cellular or culture material. In other embodiments fusion polypeptides may be directly synthesised and combined with polyhedrin in host cells or under cell free conditions that allow the formation of polyhedrin-antigen complexes and folding and production of polyhedra or polyhedra-like particles. In some embodiments, the methods increase the half-life or shelf life (stability) of an antigen prepared according to the above method or a composition comprising same. In some embodiments, the methods increase the resistance of the antigen preparation to enzymatic degradation or degradation under certain physiological or environmental conditions. In some embodiments, kits such as immunodiagnostic or immunoscreening kits comprising the isolated or purified complexes or fusion proteins and/or antibodies thereto are contemplated. In some embodiments, antibodies are produced according to a method comprising administering to a non human subject an effective amount of a complex comprising (a) a fusion polypeptide comprising a polyhedrin targeting peptide and an antigen of a pathogen or other antigen associated with a condition against which an antibody is sought; and (b) polyhedrin, wherein administration is for a time and under conditions sufficient for the antigen to induce an antibody response. In other embodiments, the fusion polypeptide is administered. In some embodiments, antibodies are used in the manufacture of a chimeric, deimmunised, humanised or human antibodies as known in the art. In another embodiment, the present invention contemplates methods for screening putative interacting (binding) agents for those that bind to a subject antigen in the form of a complex comprising polyhedrin or a fusion polypeptide as described herein. In some embodiments, the methods comprise contacting a purified complex or fusion protein of the present invention with a putative interacting agent and determining binding relative to controls. In some embodiments, binding agents are further tested for their ability to reduce the level or activity of a pathogen or cancerous cell from which the antigen is derived. Further embodiments are directed to a nucleic acid molecule encoding the fusion polypeptides described herein, host cells comprising the subject complexes or fusion polypeptides, and compositions comprising purified recombinant or modified polyhedra. Compositions may include agents to facilitate destabilisation (such as pH modifiers) or stabilisation (such as cross-linking) of the complex in vivo. Pharmaceutical compositions comprising the subject polyhedrin-antigen complexes or polyhedrin targeting peptide-antigen fusion polypeptides, or an antibody determined thereby that specifically recognises the antigen are provided. The above summary is not and should not be seen in any way as an exhaustive recitation of all embodiments of the present invention. Some figures contain colour representations or entities. Coloured versions of the figures are available from the Patentee upon request or from an appropriate Patent Office. A fee may be imposed if obtained from a Patent Office. The subject invention is not limited to particular screening procedures, specific formulations and various medical methodologies, as such may vary. Unless defined otherwise, all technical and scientific terms used herein have the same meanings as commonly understood by one of ordinary skill in the art to which this invention belongs. Any materials and methods similar or equivalent to those described herein can be used to practise or test the present invention. Practitioners are particularly directed to Ream et al., eds., Reference herein to a virus or viral antigen includes without limitation a virus or antigen therefrom from any virus family. Non-limiting examples of viral families include Adenoviridae, African swine fever-like viruses. Arenaviridae, Arterivirus, Astroviridae, Baculoviridae, Birnaviridae, Bunyaviridae, Caliciviridae, Circoviridae, Coronaviridae, Deltavirus, Filoviridae, Flaviviridae, Hepadnaviridae, Hepeviridae, Herpesviridae, Orthoinyxoviridae, Paranmyxoviridae, Picornaviridae, Poxyviridae, Reoviridae, Retroviridae and Rhabdoviridae. Particular viruses are from Paramyxoviridae, Retroviridae and Filoviridae. In some embodiments, a virus includes a virus selected from influenza virus, respiratory syncytial virus (RSV), chlamydia, adenovirdiae, mastadenovirus, aviadenovirus, herpesviridae, herpes simplex virus 1, herpes simplex virus 2, herpes simplex virus 5, herpes simplex virus 6, leviviridae, levivirus, enterobacteria phase MS2, allolevirus, poxyiridae, chordopoxyirinae, parapoxvirus, avipoxvirus, capripoxvirus, leporiipoxvirus, suipoxvirus, molluscipoxvirus, entomopoxvirinae, papovavaridae, polyomuvirus, papillomavirus, paramyxoviridae, paramyxovirus, parainfluenza virus 1, mobillivirus, measles virus, rubulavirus, mumps virus, pneumonovirinae, pneumovirus, metapneumovirus, avian pneumovirus, human metapneumovirus, picornaviridae, enterovirus, rhinovirus, hepatovirus, human hepatitis A virus, cardiovirus, andapthovirus, reoviridae, orthoreovirus, orbivirus, rotavirus, cypovirus, fijivinrus, phytoreovirus, oryzavirus, retroviridae, mammalian type B retroviruses, mammalian type C retroviruses, avian type C retroviruses, type D retrovirus group, BLV-HTLV retroviruses, lentivirus, human immunodeficiency virus I, human immunodeficiency virus 2, spumavirus, flaviviridae, hepatitis C virus, hepadnaviridae, hepatitis B virus, togaviridae, alphavirus sindbis virus, rubivirus, rubella virus, rhabdoviridae, vesiculovirus, lyssavirus, ephemerovirus, cytorhabdovirus, necleorhabdovirus, arenaviridae, arenavirus, lymphocytic choriomeningitis virus, Ippy virus, lassa virus, coronaviridae, coronavirus and torovirus. Illustrative viral pathogens include HIV, HSV, chlamydia, SARS, RSV, Dengue virus and Influenza. Another illustrative pathogen is an apicomplexal parasite such as In particular embodiments, the antigen is a polypeptide or peptide proposed to engender or facilitate the production of an effective immune response in at least some subjects. Without being bound by any particular theory or mode of action, it is proposed that the present complexes stabilise and or protect the three dimensional structure of the antigen and provide improved vehicles for effective immune response production, for antibody and in some embodiments neutralising antibody production and for immune response and antibody screening. In preparing antibodies for diagnosis or screening, an effective immune response is generally one that producing antibodies of sufficient affinity to be useful reagents in standard protocols employing antibodies, such as ELISA, RIA, RAPID, etc. In some embodiments, the antigen is recognised in the art as useful or potentially useful for generating a protective or neutralising immune response. A range of illustrative known target antigens are described herein. In other embodiments, the invention permits the characterisation of new useful antigens and conformational epitopes recognised, for example, by neutralising antibodies from infected subjects. Any viral or non-viral antigen of a pathogen or cancer may be engineered using the methods described or referenced in this specification. An “antigen” or “immunogen” or “antigenic” or “immunogenic” refers to a molecule containing one or more epitopes (either linear, conformational or both) that will stimulate an immune system to make a humoral and/or cellular antigen-specific response. Generally, a B-cell epitope will include at least about 5 amino acids but can be as small as 3-4 amino acids. A T-cell epitope, such as a cytolytic T-cell (CTL) epitope, will include at least about 7-9 amino acids, and a helper T-cell epitope at least about 12-20 amino acids. Normally, an epitope will include between about 7 and 15 amino acids, such as, 9, 10, 12 or 15 amino acids. The term “antigen” denotes both subunit antigens, (i.e., antigens which are separate and discrete from a whole organism with which the antigen is associated in nature), as well as, killed, attenuated or inactivated bacteria, viruses, fungi, parasites or other microbes. Antibodies such as anti-idiotype antibodies, or fragments thereof, and synthetic peptide mimotopes, which can mimic an antigen or antigenic determinant, are also captured under the definition of antigen as used herein. The “antigen” may comprise one or more epitopes of one or more species, subspecies, types, clades, variants, isolates, etc. and/or one or more pathogens and/or one or more cancer antigens. In some embodiments, reference to “antigen” does not include human or mammalian antigens encoded by a nucleic acid molecule expressed in humans, other than tumor antigens. In some embodiments “antigen” does not include antigens encoded by indigenous nucleic acid molecules expressed in humans. Illustrative antigens include those selected from influenza virus haemagglutinin, human respiratory syncytial virus G glycoprotein, core protein, matrix protein or other protein of Dengue virus, measles virus haemagglutinin, herpes simplex virus type 2 glycoprotein gB, poliovirus I VP1, envelope or capsid glycoproteins of HIV-I or HIV-II, hepatitis B surface antigen, diptheria toxin, Illustrative cancer antigens include KS ¼ pan-carcinoma antigen, ovarian carcinoma antigen (CA125), prostatic acid phosphate, prostate specific antigen, melanoma-associated antigen p97, melanoma antigen gp75, high molecular weight melanoma antigen (HMW-MAA), prostate specific membrane antigen, carcinoembryonic antigen (CEA), polymorphic epithelial mucin antigen, human milk fat globule antigen, colorectal tumor-associated antigens, CEA, TAG-72, LEA, Burkitt's lymphoma antigen-38.13, CD9, human B lymphoma antigen-CD20, CD33, melanoma specific antigens, ganglioside GD2, ganglioside GD3, ganglioside GM2, ganglioside GM3, tumor specific transplantation type of cell-surface antigen (TSTA), virally-induced tumor antigens, T-antigen DNA tumor viruses, envelope antigens of RNA tumor viruses, oncofetal antigen-alpha-fetoprotein, CEA of colon, bladder tumor oncofetal antigen, differentiation antigen, human lung carcinoma antigen L6, L20, antigens of fibrosarcoma, human leukemia T cell antigen-Gp37, neoglycoprotein, sphingolipids, breast cancer antigen, EGFR (Epidermal growth factor receptor), HER2 antigen, polymorphic epithelial mucin, malignant human lymphocyte antigen-APO-1, differentiation antigen, including I antigen found in fetal erythrocytes, primary endoderm, I antigen found in adult erythrocytes, preimplantation embryos. I (Ma) found in gastric adenocarcinomas, M8, M39 found in breast epithelium, SSEA-1 found in mycloid cells, VEP8, VEP9, Myl, VIM-DS, Du56-22 found in colorectal cancer, TRA-1-85 (blood group H), C14 found in colonic adenocarcinoma, F3 found in lung adenocarcinoma, AH6 found in gastric cancer, Y hapten, LeY found in embryonal carcinoma cells, TL5 (blood group A), EGF receptor found in A431 cells, El series (blood group B) found in pancreatic cancer, FC10, 2 found in embryonal carcinoma cells, gastric adenocarcinoma antigen. CO-514 (blood group Lea) found in Adenocarcinoma, NS-10 found in adenocarcinomas, CO-43 (blood group Leb), G49 found in EGF receptor of A431 cells. MH2 (blood group ALeb/Ley) found in colonic adenocarcinoma. 19.9 found in colon cancer, gastric cancer mucins, TsA7 found in myeloid cells, R24 found in melanoma, 4.2, GD3, D1.1, OFA-1, GM2, OFA-2, GD2, and M1:22:25:8 found in embryonal carcinoma cells, and SSEA-3 and SSEA-4 found in 4 to 8-cell stage embryos. Non-viral pathogens and antigens further include those from pathogenic or non-pathogenic fungi, including parasites, including apicomplexa, or uni cellular parasites, nematodes, trematodes, cestodes and plant pathogen or parasitic bacteria. In an illustrative embodiment, one important group of pathogens is the primary systemic fungal pathogens of man such In some embodiments, the pathogen is a microbe including a bacterium, fungus, virus, algae, parasite, (including ecto- or endo-parasites) prion, oomycetes, slime, moulds, nematode, mycoplasma and the like. By way of non limiting example, the microbe is selected from one or more of the following orders, genera or species: Reference herein to “bound” includes covalent and non covalent bonds. In illustrated embodiments, the bond is a covalent bond, such as between linear components of a fusion protein. Another covalent bond is a disulphide base. “Fused” refers to a covalent bond. “Synthetic” sequences, as used herein, include polynucleotides whose expression has been optimized as described herein, for example, by codon substitution, deletions, replacements and/or inactivation of inhibitory sequences usually in order to optimize expression. “Wild type” or “native” or “naturally occurring” sequences, as used herein, refers to polypeptide encoding sequences that are essentially as they are found in nature. Recombinant polypeptides and antigens can be conveniently prepared using standard protocols as described for example in Sambrook, et al., 1989 (supra), in particular Sections 16 and 17; Ausubel et al., 1994 (supra), in particular Chapters 10 and 16; and Coligan et al., Pharmaceutical compositions are conveniently prepared according to conventional pharmaceutical compounding techniques. See, for example, Remington's Pharmaceutical Sciences, 18th Ed., Mack Publishing, Company, Easton, Pa., U.S.A., 1990. The composition may contain the active agent or pharmaceutically acceptable salts of the active agent. These compositions may comprise, in addition to one of the active substances, a pharmaceutically acceptable excipient, carrier, buffer, stabilizer or other materials well known in the art. Such materials should be non-toxic and should not interfere with the efficacy of the active ingredient. The carrier may take a wide variety of forms depending on the form of preparation desired for administration, e.g. intravenous, oral or parenteral. A “pharmaceutically acceptable carrier” and/or a diluent is a pharmaceutical vehicle comprised of a material that is not otherwise undesirable i.e., it is unlikely to cause a substantial adverse reaction by itself or with the active agent. Carriers may include all solvents, dispersion media, coatings, antibacterial and antifungal agents, agents for adjusting tonicity, increasing or decreasing absorption or clearance rates, buffers for maintaining pH, chelating agents, membrane or barrier crossing agents. A pharmaceutically acceptable salt is a salt that is not otherwise undesirable. The agent or composition comprising the agent may be administered in the form of pharmaceutically acceptable non-toxic salts, such as acid addition salts or metal complexes. For oral administration, the compounds can be formulated into solid or liquid preparations such as capsules, pills, tablets, lozenges, powders, suspensions or emulsions. In preparing the compositions in oral dosage form, any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents, suspending agents, and the like in the case of oral liquid preparations (such as, for example, suspensions, elixirs and solutions); or carriers such as starches, sugars, diluents, granulating agents, lubricants, binders, disintegrating agents and the like in the case of oral solid preparations (such as, for example, powders, capsules and tablets). Because of their ease in administration, tablets and capsules represent the most advantageous oral dosage unit form, in which case solid pharmaceutical carriers are obviously employed. Tablet may contain a hinder such as tragacanth, corn starch or gelatin; a disintegrating agent, such as alginic acid; and a lubricant, such as magnesium stearate. If desired, tablets may be sugar-coated or enteric-coated by standard techniques. The active agent can be encapsulated to make it stable to passage through the gastrointestinal tract. See for example, International Patent Publication No. WO 96/11698. For parenteral administration, the composition may be dissolved in a carrier and administered as a solution or a suspension. When the agents are administered intrathecally, they may also be dissolved in cerebrospinal fluid. For transmucosal or transdermal (including patch) delivery, appropriate penetrants known in the art are used for delivering the subject complexes. For inhalation, delivery uses any convenient system such as dry powder aerosol, liquid delivery systems, air jet nebulizers, propellant systems. For example, the formulation can be administered in the form of an aerosol or mist. The agents may also be delivered in a sustained delivery or sustained release format. For example, biodegradable microspheres or capsules or other polymer configurations capable of sustained delivery can be included in the formulation. Formulations can be modified to alter pharmacokinetics and biodistribution. For a general discussion of pharmacokinetics, see, e.g., Remington's. In some embodiments the formulations may be incorporated in lipid monolayers or bilayers such as liposomes or micelles. Targeting therapies known in the art may be used to deliver the agents more specifically to certain types of cells or tissues such as, without limitation, antigen presenting cells. The actual amount of active agent administered and the rate and time-course of administration will depend on the nature and severity of the disease or condition. Prescription of treatment, e.g. decisions on dosage, timing, etc. is within the responsibility of general practitioners or specialists and typically takes into account the condition of the individual patient, the site of delivery, the method of administration and other factors known to practitioners. Examples of techniques and protocols can be found in Remington's Pharmaceutical Sciences (supra). Sustained-release preparations that may be prepared are particularly convenient for inducing immune responses. Examples of sustained-release preparations include semipermeable matrices of solid hydrophobic polymers containing the antagonist, which matrices are in the form of shaped articles, e.g., films, or microcapsule. Examples of sustained-release matrices include polyesters, hydrogels (for example, poly(2-hydroxyethyl-methacrylate), or poly(vinylalcohol)), polylactides, copolymers of L-glutamic acid and ethyl-L-glutamate, non-degradable ethylene-vinyl acetate, degradable lactic acid-glycolic acid copolymers, and poly-D-(−)-3-hydroxybutyric acid. While polymers such as ethylene-vinyl acetate and lactic acid-glycolic acid enable release of molecules for over 100 days, certain hydrogels release proteins for shorter time periods. Liposomes may be used which are of the small (about 200-800 Angstroms) unilamellar type in which the lipid content is greater than about 30% cholesterol, the selected proportion being adjusted for the optimal therapy. Stabilization of proteins may be achieved by modifying sulfhydryl residues, lyophilizing from acidic solutions, controlling moisture content, using appropriate additives, and developing specific polymer matrix compositions. The in vivo half life of proteins may be extended using techniques known in the art, including, for example, by the attachment of other elements such as polyethyleneglycol (PEG) groups. Prime-boost immunisation strategies as disclosed in the art are clearly contemplated. See for example International Publication No. WO/2003/047617. Thus, compositions may be in the form of a vaccine, priming or boosting agent. Instead of administering the protein complex directly, they could be produced in a host cell or an introduced cell, e.g. in a viral vector or in a cell based delivery system. The vector could be targeted to elements of the immune system. A cell based delivery system is designed to be implanted in a patient's body at a desired target site and contains coding sequences for the subject fusion polypeptides, complexes and polyhedra. Alternatively, the agent could be administered in a precursor form for conversion to the active form by an activating agent produced in, or targeted to, the cells to be treated. In further describing the various applications of the subject compositions in eliciting immune responses, the compositions is generally administered in an effective amount and for a time an under conditions sufficient to elicit an immune response. The compositions of the present invention may be administered as a single dose. Alternatively, the compositions may involve repeat doses or applications. The terms “effective amount” including a “therapeutically effective amount” and “prophylactically effective amount” as used herein mean a sufficient amount a composition comprising a complex as defined herein, or a cell or antibody as described herein, which provides the desired therapeutic or physiological effect and is an amount sufficient to achieve a biological effect such as to induce enough humoral or cellular immunity. Desired biological effects include but are not limited to reduced or no symptoms, remission, reduced pathogen titres, reduced vascular or cerebral compromise, reduced nasal secretions, fever etc. Undesirable effects, e.g. side effects, may sometimes manifest along with the desired therapeutic effect; hence, a practitioner balances the potential benefits against the potential risks in determining an appropriate “effective amount”. The exact amount of agent required will vary from subject to subject, depending on the species, age and general condition of the subject, mode of administration and the like. Thus, it may not be possible to specify an exact “effective amount” However, an appropriate “effective amount” in any individual case may be determined by one of ordinary skill in the art using routine experimentation. One of ordinary skill in the art would be able to determine the required amounts based on such factors as prior administration of agents, the subject's size, the severity of the subject's symptoms, pathogen load, and the particular composition or route of administration selected. The terms “treatment” or “prophylaxis” or “therapy” are used interchangeably in their broadest context and include any measurable or statistically significant amelioration in at least some subjects in one or more symptoms of a condition to be treated or in the risk of developing a particular condition. Prophylaxis may be considered as reducing the severity or onset of a condition or signs of a condition. Treatment may also reduce the severity of existing conditions. The administration of a vaccine composition is generally for prophylactic purposes. In some embodiments, a vaccine or composition of the present invention is physiologically effective if its presence results in a detectable change in the physiology of a recipient patient that enhances or indicates an enhancement in at least one primary or secondary humoral or cellular immune response against at least one strain of an pathogen or virus. In some embodiments the vaccine composition is administered to protect against infection by a pathogen. The “protection” need not be absolute, i.e., the infection need not be totally prevented or eradicated, if there is a statistically significant improvement compared with a control population or set of patients. Protection may be limited to reducing the severity or rapidity of onset of symptoms of the viral or other pathogen infection, or the development of cancer or other condition as described herein. In one embodiment, a vaccine composition of the present invention is provided to a subject either before the onset of infection (so as to prevent or attenuate an anticipated infection) or after the initiation of an infection, and thereby protects against viral infection. In some embodiments, a vaccine composition of the present invention is provided to a subject before or after onset of infection, to reduce viral transmission between subjects. It will be further appreciated that compositions of the present invention can be administered as the sole active pharmaceutical agent, or used in combination with one or more agents to treat or prevent pathogen infections or symptoms associated with such infection. The pharmaceutical composition is contemplated to exhibit therapeutic activity when administered in an amount that depends upon the particular case. The variation depends, for example, on the human or animal and the agent chosen. A broad range of doses may be applicable. Considering a subject, for example, from about 0.1 μg to 1 μg (i.e., including 0.1 μg, 0.2 μg, 0.3 μg, 0.4 μg, 0.5 μg, 0.6 μg, 0.7 μg, 0.8 μg and 0.9 μg) 0.5 μg to 50 μg, 1 μg to 10 μg, 2 μg to 200 μg, 0.1 mg to 1.0 mg (i.e., including 0.1 mg, 0.2 mg, 0.3 mg, 0.4 mg, 0.5 mg, 0.6 mg, 0.7 mg, 0.8 mg and 0.9 mg), from about 15 mg to 35 mg, about 1 mg to 30 mg or from 5 to 50 mg, or from 10 mg to 100 mg of agent may be administered per kilogram of body weight per day or per every other day or per week or per month. Therapeutic including prophylactic compositions may be administered at a dosage of about 0.1 to 20 mg/kg however dosages above or below this amount are contemplated in the ranges set out above. Dosage regimes may be adjusted to provide the optimum therapeutic response. For example, several divided doses may be administered daily, weekly, monthly or other suitable time intervals or the dose may be proportionally reduced as indicated by the exigencies of the situation. It is also possible to administer compositions in sustained release formulations. Pharmaceutical preparations are conveniently provided in unit dosage form such as tablets, capsules, powders etc. The compositions, complexes, antibodies and cells may be administered in a convenient manner such as by the oral, intravenous, intraperitoneal, intramuscular, subcutaneous, intradermal, intrathecal or suppository routes or implanting (e.g. using slow release molecules). Administration may be systemic or local. References to systemic include intravenous, intraperitoneal, subcutaneous injection, infusion as well as administration via oral, rectal, vaginal and nasal routes or via inhalation. Other contemplated routes of administration are by patch, cellular transfer, implant, sublingually, intraocularly, topically or transdermally. In some embodiments, oral or nasal administration is contemplated. Capillaries have a diameter or approximately 5 μm permitting administration of complexes that are smaller than about 1 μm diameter. Polyhedra of more than 5 μm may be administered subcutaneously or intra muscularly or by other convenient route known in the art. Polyhedra can routinely be separated based upon size. Functional variants and derivatives include “biologically active portion” or “biologically active part” or “functional part or portion” by which is meant a portion of a full-length targeting polypeptides which portion retains the activity of the full length molecule at least in so far as it retains the structural and functional abilities to target an antigen to polyhedrin. As used herein, the term “biologically active portion” includes deletion mutants and peptides, for example of at least about 20 to 200 amino acids, such as 20, 21, 22, 23, 24, 25, 30, 40, 50, 60, 70, 80, 90, 100, 120, 150, 300, 350 contiguous amino acids (and every integer in between), which retains activity. Portions of this type may be obtained through the application of standard recombinant nucleic acid techniques or synthesized using conventional or state of the art liquid or solid phase synthesis techniques. For example, reference may be made to solution synthesis or solid phase synthesis as described, for example, in Chapter 9 entitled By “derivative” is meant a polypeptide that has been derived from the basic sequence by modification, for example by conjugation or complexing with other chemical moieties or by post-translational modification techniques as would be understood in the art. The term “derivative” also includes within its scope alterations that have been made to a targeting polypeptide including additions, or deletions that provide for functionally equivalent molecules. A “part” or “portion” of a polynucleotide or polypeptide is defined as having a minimal size of at least about 20 nucleotides or amino acids and may have a minimal size of at least about 100 nucleotides or amino acids. This definition includes all sizes in the range of 10-nucleotides or amino acids including 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 or 35 nucleotides or amino acids as well as greater than 100 nucleotides or amino acids including 300, 500, 600 nucleotides or amino acids or molecules having any number of nucleotides or amino acids within these values. Reference herein to “functional variants” of targeting polypeptides or peptides or polyhedrin polypeptides include naturally or non-naturally occurring functional variants, biologically active parts or portions, precursors, derivatives, analogs and recombinant or synthetic forms having a degree of sequence similarity or the omission of one or more biologically active parts or portions sufficient to retain the functional and structural ability of the sequences identified herein to form complexes with polyhedrin as described herein. Functional variants are described further in the detailed description. The term “sequence identity” as used herein refers to the extent that sequences are identical on a nucleotide-by-nucleotide basis or an amino acid-by-amino acid basis over a window of comparison. Thus, a “percentage of sequence identity” is calculated by comparing two optimally aligned sequences over the window of comparison, determining the number of positions at which the identical nucleic acid base (e.g. A, T, C, G, I) or the identical amino acid residue (e.g., Ala, Pro, Ser, Thr, Gly, Val, Leu, Ile, Phe, Tyr, Trp, Lys, Arg, His, Asp, Glu, Asn, Gln, Cys and Met) occurs in both sequences to yield the number of matched positions, dividing the number of matched positions by the total number of positions in the window of comparison (i.e., the window size), and multiplying the result by 100 to yield the percentage of sequence identity. For the purposes of the present invention, “sequence identity” will be understood to mean the “match percentage” calculated by an appropriate method. For example, sequence identity analysis may be carried out using the DNASIS computer program (Version 2.5 for windows; available from Hitachi Software engineering Co., Ltd., South San Francisco, Calif., USA) using standard defaults as used in the reference manual accompanying the software. Terms used to describe sequence relationships between two or more polynucleotides or polypeptides include “reference sequence”, “comparison window”, “sequence identity”, “percentage of sequence identity” and “substantial identity”. A “reference sequence” is at least 12 but frequently 15 to 18 and often at least 25 monomer units, inclusive of nucleotides and amino acid residues, in length. Because two polynucleotides may each comprise (1) a sequence (i.e., only a portion of the complete polynucleotide sequence) that is similar between the two polynucleotides, and (2) a sequence that is divergent between the two polynucleotides, sequence comparisons between two (or more) polynucleotides are typically performed by comparing sequences of the two polynucleotides over a “comparison window” to identify and compare local regions of sequence similarity. A “comparison window” refers to a conceptual segment of at least 6 contiguous positions, usually about 50 to about 100, more usually about 100 to about 150 in which a sequence is compared to a reference sequence of the same number of contiguous positions after the two sequences are optimally aligned. The comparison window may comprise additions or deletions (i.e., gaps) of about 20% or less as compared to the reference sequence (which does not comprise additions or deletions) for optimal alignment of the two sequences. Optimal alignment of sequences for aligning a comparison window may be conducted by computerized implementations of algorithms (GAP, BESTFIT, FASTA, and TFASTA in the Wisconsin Genetics Software Package Release 7.0, Genetics Computer Group, 575 Science Drive Madison, Wis., USA) or by inspection and the best alignment (i.e., resulting in the highest percentage homology over the comparison window) generated by any of the various methods selected. Reference also may be made to the BLAST family of programs as for example disclosed by Altschul et al. The term “recombinant” may be used herein to describe a nucleic acid molecule and means a polynucleotide of genomic, cDNA, semisynthetic, or synthetic origin which, by virtue of its origin or manipulation: (1) is not associated with all or a portion of the polynucleotide with which it is associated in nature; and/or (2) is linked to a polynucleotide other than that to which it is linked in nature. The term “recombinant” as used with respect to a protein or polypeptide means a polypeptide produced by expression of a recombinant polynucleotide. “Recombinant host cells,” “host cells,” “cells,” “cell lines,” “cell cultures,” and other such terms denoting prokaryotic microorganisms or eukaryotic cell lines cultured as unicellular entities, are used interchangeably, and refer to cells which can be, or have been, used as recipients for recombinant vectors or other transfer DNA, and include the progeny of the original cell which has been transfected. “Hybridization” or “hybridize” is used herein to denote the pairing of complementary nucleotide sequences to produce a DNA-DNA hybrid or a DNA-RNA hybrid. Hybridization can occur under varying circumstances as known to those of skill in the art. The phrase “hybridizing specifically to” and the like refer to the binding, duplexing, of hybridizing of a molecule only to a particular nucleotide sequence under stringent conditions as known in the art. The terms “antibody” and “antibodies” include polyclonal and monoclonal antibodies and all the various forms derived from monoclonal antibodies, including but not limited to full-length antibodies (e.g. having an intact Fc region), antigen-binding fragments, including for example, Fv, Fab, Fab′ and F(ab′)2 fragments; and antibody-derived polypeptides produced using recombinant methods such as single chain antibodies. The terms “antibody” and “antibodies” as used herein also refer to human antibodies produced for example in transgenic animals or through phage display, as well as subject antibodies, santibodies, primatised antibodies or deimmunised antibodies. It also includes other forms of antibodies that may be therapeutically acceptable and antigen-binding fragments thereof, for example single domain antibodies derived from cartilage marine animals or Camelidae, or from libraries based on such antibodies. The selection of fragment or modified forms of the antibodies may also involve any effect the fragments or modified forms have on their half-lives. The term “monoclonal antibody” is used herein to refer to an antibody obtained from a population of substantially homogeneous antibodies. That is, the individual antibodies comprising the population are identical except for naturally occurring mutations that may be present in minor amounts. The term “monoclonal” as used herein indicates the character of the antibody as being obtained from a substantially homogeneous population of antibodies, and is not used to indicate that the antibody was produced by a particular method. For example, monoclonal antibodies in accordance with the present invention may be made by the hybridoma method described by Kohler and Milstein, Vectors available for cloning and expression in host cell lines are well known in the art, and include but are not limited to vectors for cloning and expression in mammalian cell lines, vectors for cloning and expression in bacterial cell lines, vectors for cloning and expression in phage and vectors for cloning and expression insect cell lines. The antibodies can be recovered using standard protein purification methods. Chemical analogs of antigens or polyhedrin molecules may be routinely employed where appropriate. Analogs contemplated herein include, but are not limited to, modifications of side chains, incorporation of unnatural amino acids and/or their derivatives during peptide, polypeptide or protein synthesis and the use of crosslinkers and other methods which impose conformational constraints on the proteinaceous molecule or their analogs. The Invention provides a method for producing an antibody comprising immunising a non-human animal or screening expression products of a library of human immunoglobulin genes with a fusion or complex protein or polyhedra as described herein, or a nucleic acid encoding same and isolating an antibody that binds specifically to the subject antigen or to all or part of a pathogen or tissue comprising same. In another embodiment, the invention provides an antibody produced by the methods described herein using a subject protein or complex or a subject, human or humanised form thereof. The antibody is preferable monoclonal rather than polyclonal and is preferably subject, humanised, deimmunised or is a human antibody. Reference to functional variants include those that are distinguished from a naturally-occurring form or from forms presented herein by the addition, deletion and/or substitution of at least one amino acid residue. Thus, variants include proteins derived from the native protein by deletion (so-called truncation) or addition of one or more amino acids to the N-terminal and/or C-terminal end of the native protein; deletion or addition of one or more amino acids at one or more sites in the native protein; or substitution of one or more amino acids at one or more sites in the native protein. Variant proteins encompassed by the present invention are biologically active, that is, they continue to possess the desired biological activity of the parent protein (e.g., immunogenicity or ability to form complexes with polyhedrin or encapsulate at least partially the antigen of interest). Variants may result from, for example, genetic polymorphism or from human manipulation. Biologically active variants of a viral polypeptide will typically have at least 40%, 50%, 60%, 70%, generally at least 75%, 80%, 85%, preferably about 90% to 95% or more, and more preferably about 98% or more sequence similarity or identity with the published amino acid sequence for the protein described herein as determined by sequence alignment programs described elsewhere herein using default parameters. In some embodiments, percentage identified refers to the full length polypeptide or to the parent molecule from which the variant is derived. A biologically active variant of a subject polypeptide may differ from that polypeptide generally by as much 100, 50 or 20 amino acid residues or suitably by as few as 1-15 amino acid residues, as few as 1-10, such as 6-10, as few as 5, as few as 4, 3, 2, or even 1 amino acid residue. A variant polypeptide may be altered in various ways including amino acid substitutions, deletions, truncations, and insertions. Methods for such manipulations are generally known in the art. For example, amino acid sequence variants of a subject polypeptide can be prepared by mutations in the DNA. Methods for mutagenesis and nucleotide sequence alterations are well known in the art. See, for example, Kunkel, Variant subject polypeptides may contain conservative amino acid substitutions at various locations along their sequence, as compared to the reference amino acid sequence. A “conservative amino acid substitution” is one in which the amino acid residue is replaced with an amino acid residue having a similar side chain. Families of amino acid residues having similar side chains have been defined in the art, which can be generally sub-classified as follows: Acidic: The residue has a negative charge due to loss of H ion at physiological pH and the residue is attracted by aqueous solution so as to seek the surface positions in the conformation of a peptide in which it is contained when the peptide is in aqueous medium at physiological pH. Amino acids having an acidic side chain include glutamic acid and aspartic acid. Basic: The residue has a positive charge due to association with H ion at physiological pH or within one or two pH units thereof (e.g., histidine) and the residue is attracted by aqueous solution so as to seek the surface positions in the conformation of a peptide in which it is contained when the peptide is in aqueous medium at physiological pH. Amino acids having a basic side chain include arginine, lysine and histidine. Charged: The residues are charged at physiological pH and, therefore, include amino acids having acidic or basic side chains (i.e., glutamic acid, aspartic acid, arginine, lysine and histidine). Hydrophobic: The residues are not charged at physiological pH and the residue is repelled by aqueous solution so as to seek the inner positions in the conformation of a peptide in which it is contained when the peptide is in aqueous medium. Amino acids having a hydrophobic side chain include tyrosine, valine, isoleucine, leucine, methionine, phenylalanine and tryptophan. Neutral/polar: The residues are not charged at physiological pH, but the residue is not sufficiently repelled by aqueous solutions so that it would seek inner positions in the conformation of a peptide in which it is contained when the peptide is in aqueous medium. Amino acids having a neutral/polar side chain include asparagine, glutamine, cysteine, histidine, serine and threonine. This description also characterizes certain amino acids as “small” since their side chains are not sufficiently large, even if polar groups are lacking, to confer hydrophobicity. With the exception of proline, “small” amino acids are those with four carbons or less when at least one polar group is on the side chain and three carbons or less when not. Amino acids having a small side chain include glycine, serine, alanine and threonine. The gene-encoded secondary amino acid proline is a special case due to its known effects on the secondary conformation of peptide chains. The structure of proline differs from all the other naturally-occurring amino acids in that its side chain is bonded to the nitrogen of the a amino group, as well as the α-carbon. Several amino acid similarity matrices (e.g., PAM120 matrix and PAM250 matrix as disclosed for example by Dayhoff et al. 1978, (supra), A model of evolutionary change in proteins. Matrices for determining distance relationships In M. O. Dayhoff, (ed.), Atlas of protein sequence and structure, Vol. 5, pp. 345-358, National Biomedical Research Foundation, Washington D.C.; and by Gonnet et al., The degree of attraction or repulsion required for classification as polar or nonpolar is arbitrary and, therefore, amino acids specifically contemplated by the invention have been classified as one or the other. Most amino acids not specifically named can be classified on the basis of known behavior. Amino acid residues can be further sub-classified as cyclic or noncyclic, and aromatic or nonaromatic, self-explanatory classifications with respect to the side-chain substituent groups of the residues, and as small or large. The residue is considered small if it contains a total of four carbon atoms or less, inclusive of the carboxyl carbon, provided an additional polar substituent is present; three or less if not. Small residues are, of course, always nonaromatic. Dependent on their structural properties, amino acid residues may fall in two or more classes. For the naturally-occurring protein amino acids, sub-classification according to this scheme is presented in the Table 1. Conservative amino acid substitution also includes groupings based on side chains. For example, a group of amino acids having aliphatic side chains is glycine, alanine, valine, leucine, and isoleucine; a group of amino acids having aliphatic-hydroxyl side chains is serine and threonine; a group of amino a ids having amide-containing side chains is asparagine and glutamine; a group of amino acids having aromatic side chains is phenylalanine, tyrosine, and tryptophan; a group of amino acids having basic side chains is lysine, arginine, and histidine; and a group of amino acids having sulphur-containing side chains is cysteine and methionine. For example, it is reasonable to expect that replacement of a leucine with an isoleucine or valine, an aspartate with a glutamate, a threonine with a serine, or a similar replacement of an amino acid with a structurally related amino acid will not have a major effect on the properties of the resulting variant polypeptide. Whether an amino acid change results in a functional subject polypeptide can readily be determined by assaying its activity. Conservative substitutions are shown in Table 2 (below) under the heading of exemplary substitutions. More preferred substitutions are shown under the heading of preferred substitutions. Amino acid substitutions falling within the scope of the invention, are, in general, accomplished by selecting substitutions that do not differ significantly in their effect on maintaining (a) the structure of the peptide backbone in the area of the substitution, (b) the charge or hydrophobicity of the molecule at the target site, or (c) the bulk of the side chain. After the substitutions are introduced, the variants are screened for biological activity Alternatively, similar amino acids for making conservative substitutions can be grouped into three categories based on the identity of the side chains. The first group includes glutamic acid, aspartic acid, arginine, lysine, histidine, which all have charged side chains; the second group includes glycine, serine, threonine, cysteine, tyrosine, glutamine, asparagine; and the third group includes leucine, isoleucine, valine, alanine, proline, phenylalanine, tryptophan, methionine, as described in Zubay, G., Thus, a predicted non-essential amino acid residue in a subject polypeptide is typically replaced with another amino acid residue from the same side chain family. Alternatively, mutations can be introduced randomly along all or part of a subject polynucleotide coding sequence, such as by saturation mutagenesis, and the resultant mutants can be screened for an activity of the parent polypeptide to identify mutants which retain that activity. Following mutagenesis of the coding sequences, the encoded peptide can be expressed recombinantly and the activity of the peptide can be determined. Accordingly, the present invention also contemplates variants of the subject polypeptides provided herein or their biologically-active fragments, wherein the variants are distinguished from the provided sequences by the addition, deletion, or substitution of one or more amino acid residues. In general, variants will display at least about 30, 40, 50, 55, 60, 65, 70, 75, 80, 85, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99% similarity to a reference subject polypeptide sequence. Desirably, variants will have at least 30, 40, 50, 55, 60, 65, 70, 75, 80, 85, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99% sequence identity to a parent subject polypeptide sequence. Moreover, sequences differing from the disclosed sequences by the addition, deletion, or substitution of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 30, 40, 50, 60, 70, 80, 90, 100 or more amino acids but which retain the biological activity of the parent subject polypeptide are contemplated. Variant subject polypeptides also include polypeptides that are encoded by polynucleotides that hybridize under stringency conditions as defined herein, especially high stringency conditions, to disclosed polynucleotide sequences, or the non-coding strand thereof. In some embodiments, variant polypeptides differ from a prior art or wild-type sequence by at least one but by less than 50, 40, 30, 20, 15, 10, 8, 6, 5, 4, 3 or 2 amino acid residue(s). In another, variant polypeptides differ from the recited sequence by at least 1% but less than 20%, 15%, 10% or 5% of the residues. (If this comparison requires alignment the sequences should be aligned for maximum similarity. “Looped” out sequences from deletions or insertions, or mismatches, are considered differences.) The differences are, suitably, differences or changes at a non-essential residue or a conservative substitution. A “non-essential” amino acid residue is a residue that can be altered from the wild type sequence of an embodiment polypeptide without abolishing or substantially altering one or more of its activities. Suitably, the alteration does not substantially alter one of these activities, for example, the activity is at least 20%, 40%, 60%, 70% or 80% of wild-type. An “essential” amino acid residue is a residue that, when altered, results in abolition of an activity of the parent molecule such that less than 20% of the parent activity is present. The present invention is further described by the following non-limiting Examples. ELISpot assay for the detection of IFN-γ A recombinant baculovirus transfer vector was constructed to encode various forms of HIV-1 Gag in frame with a nucleotide sequence of H1-helix of Bm-CPV polyhedrin (Ijiri et al., 2009 (supra)). A recombinant form of Bm-CPV (AcCP-H) which produces polyhedrin and further produces cubic polyhedra was used in this study (Mori et al, 1993 (supra)). The H1 protein functions as a polyhedrin-recognition signal and Gag-H1 protein is incorporated together with polyhedrin into polyhedra Gag MicroCubes. Polyhedra (Gag MicroCubes) were recovered and purified from the Western blot analysis showed that Gag was successfully incorporated into polyhedra. Three bands could be detected which corresponded to full-length Gag (p55), Gag lacking p6, and p39. A mutant form of Gag, where dimer formation is inhibited, was incorporated into the polyhedra crystals at a similar level compared to wild-type Gag. Using both an ELISA and Western blotting an estimated amount of 10.9 μg of Gag protein was incorporated per mg of polyhedrin protein ( Gag MicroCubes are highly stable in the presence of trypsin ( The Gag protein incorporated in MicroCubes is more stable to trypsin degradation than soluble Gag suggesting that it will provide a stable complex and sustained release of antigen when injected in vivo. To investigate immunogenicity of the HIV-1 Gag MicroCubes in vivo, compared to soluble HIV gag protein, in a dose ranging study. 6 BALB/c mice per group, immunized with 100 μl immunogen in PBS subcutaneously, at weeks 0, 4 and 8 Group A; High dose HIV gag MicroCubes (approx 450 μg, containing 5 μg HIV gag) Group B; Mid dose HIV gag MicroCubes (approx 90 μg, containing 1.0 μg HIV gag) Group C; Low dose HIV gag MicroCubes (approx 18 μg, containing 0.2 μg HIV gag) Group D; High dose HIV gag soluble protein 5 μg Group E; Mid dose HIV gag soluble protein 1.0 μg Group F; Low dose HIV gag soluble protein 0.2 μg Venous blood is collected from animals at weeks 0 (pre-bleed), 4 and 8. Animals are sacrificed at week 10 when the spleens are taken for assessment of T cell responses to the immunogens, and a terminal heart bleed is performed for serum for antibody assessments. T-cell responses are assessed in IFN-γ and IL-2 ELISPOT assays. Briefly, 5×10′ spleen cells were added to wells coated with monoclonal antibodies to either murine IFN-γ or murine IL-2. These cells were stimulated with the following antigens; media alone as negative control, Con A as positive control, HIV gag soluble protein (10, 1.0, 0.1 μg/ml), HIV Gag MicroCubes (100, 10, 1.0 μg total protein), control protein (polyhedra crystals alone), HIV Gag overlapping peptide pools I and II (see After overnight incubation cells are removed and the ELISPOTs developed. Spot forming cells/106 input splenocytes are calculated using the AID ELISPOT imaging system. Antibody responses to HIV gag was assessed using ELISA using recombinant HIV Gag soluble protein as antigen (Keoshkerian et al., Strong antibody responses to Gag were detected in both the soluble Gag and Gag MicroCube immunized mice. The maximal antibody litre of 1:25600 was observed for the 5.0 μg group with weaker responses at lower doses (see T-cell responses were assessed using IFN-γ and IL-2 ELISPOT assays. MicroCubes elicited very strong IFN-γ and IL-2 responses to Gag p55 (>500 SFC/106 cells). Slightly reduced responses were observed against the smaller fragments Gag p39 or p24. Peak IFN-γ responses were seen in the 1.0 μg group, with slightly lower responses at 5.0 μg. Importantly, higher IFN-γ responses were seen with 1.0 μg of the Gag MicroCubes (mean 210 SFC/106 cells) than with 1.0 μg gag (mean 150 SFC/106 cells). Strong IFN-γ responses to the CPV polyhedrin protein were also seen, but this was not observed in the IL-2 assay. Assays of responses to two pools of overlapping peptides corresponding to the N- and C-terminal regions of the HIV-1 Gag protein showed that the majority of the IFN-γ and IL-2 responses were directed to the second half of the protein. The robust IFN-γ and IL-2 responses to the Gag protein and Gag peptides observed after injection with Gag MicroCubes imply that this vaccine elicits both CD4 and CD8 responses without the need for adjuvant. The present invention provides a vaccine platform against infectious diseases based on ultra-stable crystals or MicroCubes (polyhedra) produced by common insect viruses. MicroCubes present two features that set them apart from existing vaccine strategies: a novel presentation of antigens as a para-crystalline array and a unique slow-release delivery mechanism. In sonic embodiments, these qualities provide immunogenicity and stability. In some embodiments, MicroCubes are used in respect of diseases that require a vigorous cellular immune response, such as HIV or malaria. In other embodiments. MicroCubes are proposed for vaccination for the developing world by offering single-shot immunizations and reducing the need for a cold chain in vaccine supply. One aspect of the invention is to employ the natural function of viral polyhedra, virus-containing crystals of the polyhedrin protein, that protect the particles of many insect viruses from environmental insults. The striking physicochemical stability of polyhedra means that the infectivity of the virus can be preserved in soil for years at ambient temperature. Recently, the much-anticipated structure of cypovirus polyhedra provided unprecedented opportunities to engineer these crystals to efficiently incorporate proteins derived from human pathogens in place of the virus particles throughout and on the surface of crystals. Herein, these crystals are referred to as “MicroCubes” owing to their shape and size (0.5-10 μm). Subunit vaccines fail to elicit a strong cellular immune response necessary to protect against a number of major pathogens (e.g. HIV). Attenuated viruses are less safe and challenging—if not impossible—to engineer for many diseases (e.g. malaria). In contrast, MicroCubes are proposed to induce both humoral and cellular responses against a range of antigens because of an improved presentation of the antigen and their particulate nature. In addition, the highly multivalent presentation of the antigen and the slow-release delivery mechanism mean that the immune responses should also be much stronger and more sustained than any available subunit vaccine, even with single-shot immunizations. To date, the advantages of symmetrical presentation of antigens have only been explored in specific examples that lack the potential of a generic vaccine platform. For instance, although very successful in the current papillomavirus vaccines (e.g. “Gardasil”) and hepatitis B vaccines (e.g. “Engerix-B”), vaccines based on virus-like particles are not generally applicable especially when large or multiple antigens are required. In contrast, in some embodiments. MicroCubes are proposed to tolerate cargoes as large as whole virus particles and even multiple different antigens at once. As shown herein, Gag MicroCubes induce a strong immune response including both a specific antibody production and a robust cell response when injected in mice. This could not be anticipated from background information as the crystals may have been rapidly cleared from the organism, or unable to be processed by antigen-presenting cells, or capable of inducing only either humoral or cellular responses. MicroCubes can be engineered to efficiently incorporate the HIV-1 Gag protein Six constructs of the Gag protein were cloned into a custom plasmid pDEST-H1 as N-terminal fusion with the Bm-CPV H1-tag. These constructs were the full-length p55 Gag protein of HIV-1 NL4.3, GagAp6 and Gag-WM (W316M317/AA mutant reducing Gag dimerization). C-terminal His6-tag fusions of each of these constructs were also engineered. Recombinant baculoviruses were obtained by cellfectin-mediated co-transfection of a modified pDEST-H1 vector and a linearised baculovirus genome (BaculoGold, Novagen). MicroCubes were produced by co-infection of Sf9 cells with the resulting high-titer baculovirus stocks and a baculovirus coding for the polyhedrin protein of Bm-CPV. MicroCubes are purified from infected cells by sonication and differential centrifugation as described in Ijiri et al., A time course revealed that Gag is incorporated into MicroCubes as early as 24 h post-infection. Incorporation levels increased at 48 h and dramatically dropped beyond 72 h post-infection (data not shown). For vaccination purposes, MicroCubes were further purified on a 45-65% (w/w) sucrose step gradient. One or two distinct bands corresponding to the crystals were observed depending on the preparation. The resulting crystals were purified to homogeneity and only the Gag and Bm-CPV polyhedrin proteins was detected even on a grossly overloaded gel as confirmed by mass spectrometry ( Co-expression with III-EGFP to produced doubly-labeled MicroCubes. Fluorescent crystals were obtained ( Highly purified crystals were shown to be sterile (NATO-accredited sterility test; Silliker Australia) and free of significant LPS contamination (<0.02 EU per injection; Strong Gag-specific, dose-escalating humoral responses were observed for soluble Gag and Gag MicroCubes as assessed by ELISA against recombinant Gag. Maximal antibody titres of 2.6×104 (soluble Gag) and 1.3×104 (Gag MicroCubes) were measured for the 5.0 μg groups ( Gag MicroCubes also elicited very strong T-cell responses measured by IFN-γ and IL-2 ELISPOT responses to Gag p55 (>200 SFC/110′ cells) and slightly weaker responses against the p39 or p24 domains of Gag. Peak IFN-γ responses were seen in the 1.0 μg group, with slightly lower responses at 5.0 μg. Higher IFN-γ responses were seen with 1.0 μg of the Gag MicroCubes (mean 205 SFC/106 cells) than with 1.0 μg soluble Gag (mean 160 SFC/106 cells) ( T-cell responses to soluble Gag were also strong and indeed comparable to those of MicroCubes, contrary to the initial hypothesis. This hypothesis assumed that unadjuvanted recombinant protein would not induce significant cellular responses. However the robust cellular and humoral responses seen here can be explained here by the particulate nature of this preparation of recombinant Gag which is known to form aggregates and VLPs in the condition of injection. In addition, slightly higher LPS levels (0.04 vs. <0.02 EU/injection for MicroCubes) were consistently observed in soluble Gag produced in In conclusion, the robust Gag-specific IFN-γ and IL-2 responses, and high titre antibody responses, observed after immunization with Gag MicroCubes demonstrate that this vaccine elicits strong cellular and humoral immunity without the need for adjuvant. This proof-of-concept provides a solid basis to investigate the magnitude of these responses in comparison with established vaccine strategies. Due to the natural robustness of polyhedra and their protective function in the viral cycle, it was hypothesized that cargoes in MicroCubes would be protected from degradation. To test this idea, MicroCubes were incubated at various temperatures and analysed by Western blot to monitor the levels and integrity of the Gag protein. Freezing at −20° C. and freeze-drying were initially investigated. Minor losses were observed and these conditions of storage were avoided in subsequent experiments. This was attributed to increase adherence to the plastic tube which was particularly obvious after freeze-dry where a white film of MicroCubes was clearly deposited on the side of the tube (data not shown). In contrast, Gag MicroCubes were found to be highly stable between 4° C. and 21° C. aid even at 37° C. A comparison with soluble Gag is presented in At 37° C., the highest temperature of this set of experiments, an intermediate situation was observed. (Gag initially appeared to be completely protected but started to degrade from day 4 and became eventually undetectable by day 14 (data not shown). Further experiments were carried out to try to identify the cause of Gag degradation in MicroCubes and try to prevent it. First, the susceptibility of Gag to proteolytic degradation was investigated. As expected, soluble Gag was found to be extremely sensitive to trypsin degradation: the incubation of 10 μg of soluble Gag at 37° C. with trypsin (10 μg/mL) resulted in complete loss of Gag in less than 10 min ( In conclusion, when embedded in MicroCubes, the (Gag protein is protected from proteolytic degradation and stable for the duration of the experiment (II weeks) between 4° C. and 21° C. Gag is also stable when incubated at 37° C. if serum is added and the overall stability of Gag MicroCubes appears very promising for a vaccine tailored for the developing world. Fine characterization of MicroCube protective capacities is investigated in using stabilising additives, different crystal formulations and incubations closer to field conditions. Antigen MicroCubes Retain their Immunogenicity after Prolonged Storage at 21° C. and Trypsin Treatment Immunogenicity studies were performed on BALB/c mice that received three subcutaneous immunizations with Gag MicroCubes (week 0, 4 and 6; 1 μg equivalent Gag) previously incubated at 4° C., 21° C. or 37° C. for a week or trypsin treated for an hour ( IFN-γ and IL-2 ELISPOT assays were used to assess in vitro the ability of naturally induced HIV-specific T cells from HIV positive subjects to recognize Gag expressed within MicroCubes. We used Peripheral Blood Mononuclear Cells from HIV positive subjects and tested them for recognition of control and Gag MicroCubes, recombinant Gag protein and overlapping peptides. Strong positive responses to Gag MicroCubes was observed in 4 out of 6 subjects who had Gag T-cell responses, as determined by positive responses to Gag proteins or peptides (data not shown). The results for two 111V positive donors (A, B) and one HIV negative donor (C) are presented in The NALP3 inflammasome recognizes crystalline material appearing in joint fluids as a danger signal. Silica and Alum crystals have recently been demonstrated to exert their inflammatory and immunogenic properties via activation of the NALP3 inflammasome (Hornung et al, Human PBMCs from several donors were incubated with purified MicroCubes. Pro-IL-1β is not constitutively expressed and requires transcriptional induction in response to e.g. a TLR stimulus. MicroCubes did not induce IL-1β cleavage and release in human PBMCs by themselves, however, LPS-primed PBMCs strongly responded to the addition of MicroCubes in a dose-dependent manner ( In order to decipher the upstream mechanisms involved in MicroCube-inducd IL-β secretion, it was tested whether or not uptake of crystalline inflammasome activators influenced cell activation. Human PBMCs were pretreated with Latrunculin A, an inhibitor of phagocytosis, which impairs actin filament assembly and subsequently stimulated with MicroCubes as well as with the non-crystalline NALP3 activator, Nigericin. Latrunculin A potently inhibited IL-1β release following MicroCubes while the response to Nigericin was unaffected ( In order to investigate whether MicroCubes can activate the NALP3 inflammasome, experiments were performed in immortalized murine macrophages from mice deficient in NALP3 or ASC (Hornung et al. supra). Macrophages from wild-type mice produced large amounts of IL-1β following treatment with descending amounts of MicroCube exposure ( Overall, these results clearly demonstrate that MicroCubes activate the ASC/NALP3 inflammasome producing mature IL1β in a phagocytosis dependent manner. The inflammasome activation may have potent proinflammatory effects in vivo which could account for at least part of the auto-adjuvant effect of MicroCube stimulation observed in the murine experiments. Many modifications will be apparent to those skilled in the art without departing from the scope of the present invention. A stable immunogenic or vaccine composition comprising a complex or polyhedra comprising same comprising an antigen of a pathogen or other antigen against which a immune response is sought in a human or non-human animal subject and a polyhedrin protein derived from a cytoplasmic polyhedrosis virus (CPV), Delivery of the complex to a subject in substantially polyhedral form induces an immune response thereto. Methods of using same to elicit an immune response. 1. A stable immunogenic or vaccine composition, comprising a complex comprising:
an antigen of a pathogen or other antigen against which an immune response is sought in a human or non-human animal subject; and a polyhedrin protein derived from a cytoplasmic polyhedrosis virus (CPV), wherein delivery of the complex to a subject in substantially particulate polyhedral form induces an immune response thereto. 2. The composition of 3. The composition of 4. The composition of 5. The composition of 6. The composition of 7. The composition of 8. The composition of 9. The composition of 10. The composition of 11. A vaccine for the treatment or prevention of an infection, disease or condition associated with antigen, comprising the composition of 12. A method for eliciting an immune response in a subject or patient, the method comprising administering to the subject or patient an effective amount of a composition of 13. A method for immunizing a subject against infection or disease or a condition associated with the antigen, comprising administering to the subject a composition of 14. A method for treating or preventing infection by a pathogen or a cancer or other condition, comprising administering to the subject a composition of 15. A method for producing an isolated or purified antibody or immune cell that specifically binds to an antigen of a pathogen or other antigen against which a immune response is sought in a human or non-human animal subject or patient, comprising administering to a subject an effective amount of a composition of FIELD
BACKGROUND
SUMMARY
BRIEF DESCRIPTION OF THE FIGURES
DETAILED DISCUSSION OF PARTICULAR EMBODIMENTS
Example 1
Material and Methods
Production of Gag Polyhedra
Purification of Polyhedra
Purification Using a Sucrose Gradient
0.45% (w/w) 9 g 11 ml 0.50% 10 g 10 ml 0.55% 11 g 9 ml 0.60% 12 g 8 ml 0.65% 13 g 7 ml
Removal of Polyhedra from Sucrose
Gag Western Blot
Murine ELISPOT Protocol
Reagents:
ELISPOT antibody pairs IFN-gamma (murine) Mabtech AN18 (rat IgG1, coating) 3321 3-1000 (1 mg) R4-6A2 (rat IgG1, detector) 3321-6-250 (250 μg) IL-2 (murine) Mabtech 1A12 (rat IgG2a) 3441-3-1000 (1 mg) 5H4 (rat IgG2b) 3441-6-250 (250 μg) IL-5 (murine) Mabtech TRFK5 (rat IgG1) 3391-3-1000 (1 mg) TRFK4 (rat IgG2a) 3391-6-250 (250 μg) IFN-gamma (rat) Mabtech rIFNg-I (mouse IgG1) 3220-3-1000 (1 mg) rIFNg-II (mouse IgG1) 3220-6-250 (250 μg) Streptavidin-alkaline Sigma S2890 phosphatase (1 mg) BCIP/NBT liquid substrate Sigma B1911 (100 ml) ELISPOT plates Millipore MSIPS4510 PBS (without Mg and Invitrogen 14190-250 Calcium) RPMI 1640, no glutamine (10 × Invitrogen 21870-092 500 ml) FCS Invitrogen 16000-044 Procedure—Example Using IFN-γ Antibody Pairs, the Same Antibody Concentrations are Used for all Antibody Pairs.
Preparation of Plates.
Addition of Splenocytes, Peptide Antigens and Peptide Pools
Plate Development.
Important.
Example 2
Baculovirus HIV-1 Gag with an N-Terminal H1 Sequence
Example 3
Production of Polyhedra Comprising HIV-1 Gag Antigen
Example 4
Immobilisation of Gag into Polyhedra
Example 5
Stable MicroCubes are Produced
Example 6
Assessment of immunogenicity of HIV Gag MicroCubes
Murine Immunogenicity
Aim
Study Design
Methods
Example 7
Discussion
Example 8
Engineering of Illustrative Antigen-MicroCubes (Polyhedra)
MicroCubes can Incorporate Two Antigens Simultaneously
Example 9
Immunogenicity Study of Antigen MicroCubes in a Marine Model
Antigen MicroCubes are Safe and Highly Immunogenic in Mice
Example 10
Distinctive Features of MicroCubes: Robustness and Self-Adjuvanting Properties
MicroCubes Protect Antigen Against Proteolytic Degradation and Heat Denaturation.
Example 11
Presentation of Antigens to Hunan T-Cells
Example 12
MicroCubes Induce Release of Mature IL-1β in Human PBMCs
Example 13
MicroCubes Activate the NALP3 Inflammasome
Amino acid sub-classification Sub-classes Amino acids Acidic Aspartic acid, Glutamic acid Basic Noncyclic: Arginine, Lysine; Cyclic: Histidine Charged Aspartic acid, Glutamic acid, Arginine, Lysine, Histidine Small Glycine, Serine, Alanine, Threonine, Proline Polar/neutral Asparagine, Histidine, Glutamine, Cysteine, Serine, Threonine Polar/large Asparagine, Glutamine Hydrophobic Tyrosine, Valine, Isoleucine, Leucine, Methionine, Phenylalanine, Tryptophan Aromatic Tryptophan, Tyrosine, Phenylalanine Residues that Glycine and Proline influence chain orientation Exemplary and Preferred Amino Acid Substitutions Original Preferred residue Exemplary substitutions substitutions Ala Val, Leu, Ile Val Arg Lys, Gln, Asn Lys Asn Gln, His, Lys, Arg Gln Asp Glu Glu Cys Ser Ser Gln Asn, His, Lys, Asn Glu Asp, Lys Asp Gly Pro Pro His Asn, Gln, Lys, Arg Arg Ile Leu, Val, Met, Ala, Phe, Norleu Leu Leu Norleu, Ile, Val, Met, Ala, Phe Ile Lys Arg, Gln, Asn Arg Met Leu, Ile, Phe Leu Phe Leu, Val, Ile, Ala Leu Pro Gly Gly Ser Thr Thr Thr Ser Ser Trp Tyr Tyr Tyr Trp, Phe, Thr, Ser Phe Val Ile, Leu, Met, Phe, Ala, Norleu Leu BIBLIOGRAPHY