Facile Synthesis of Metalloporphyrin Polymers
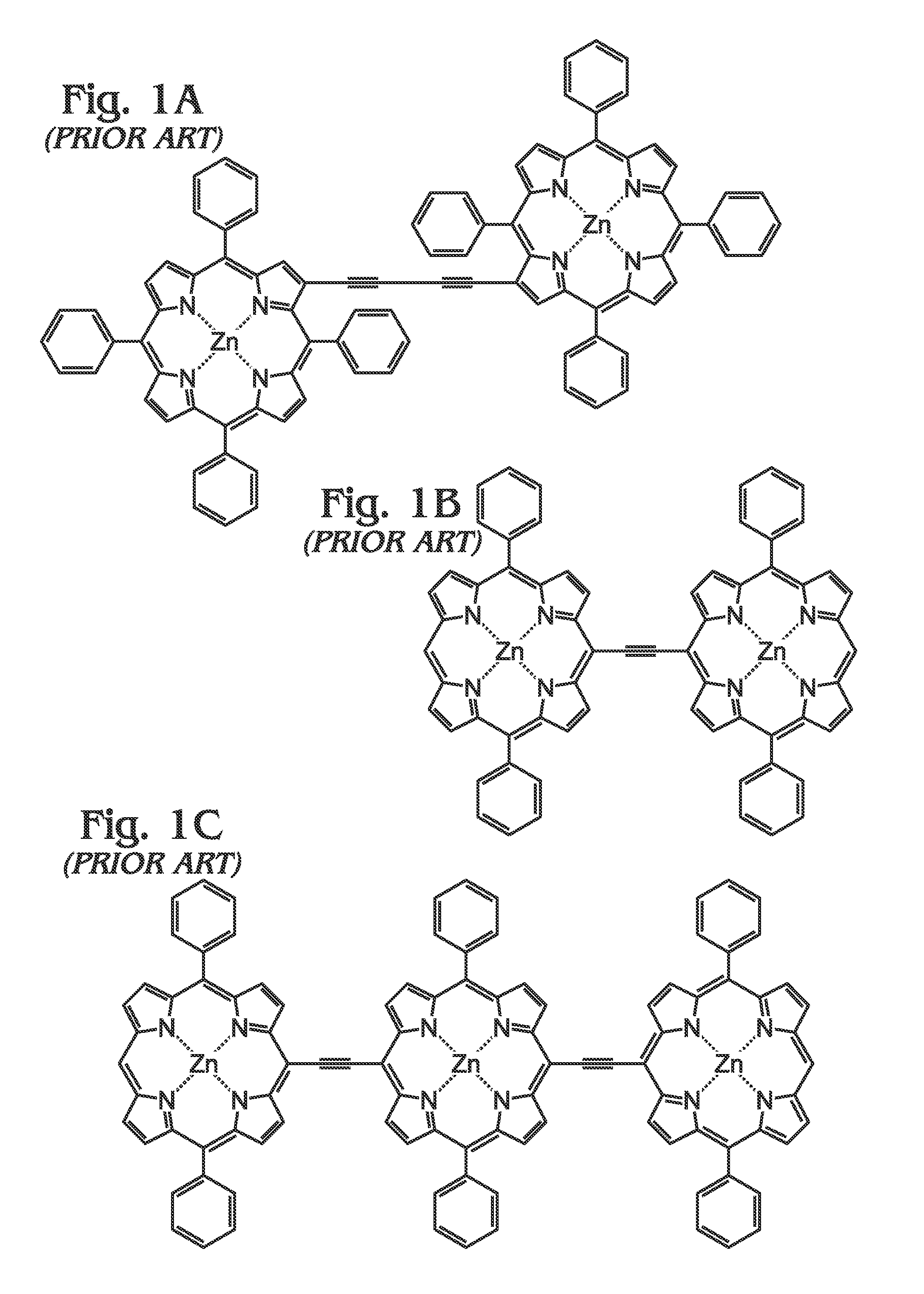
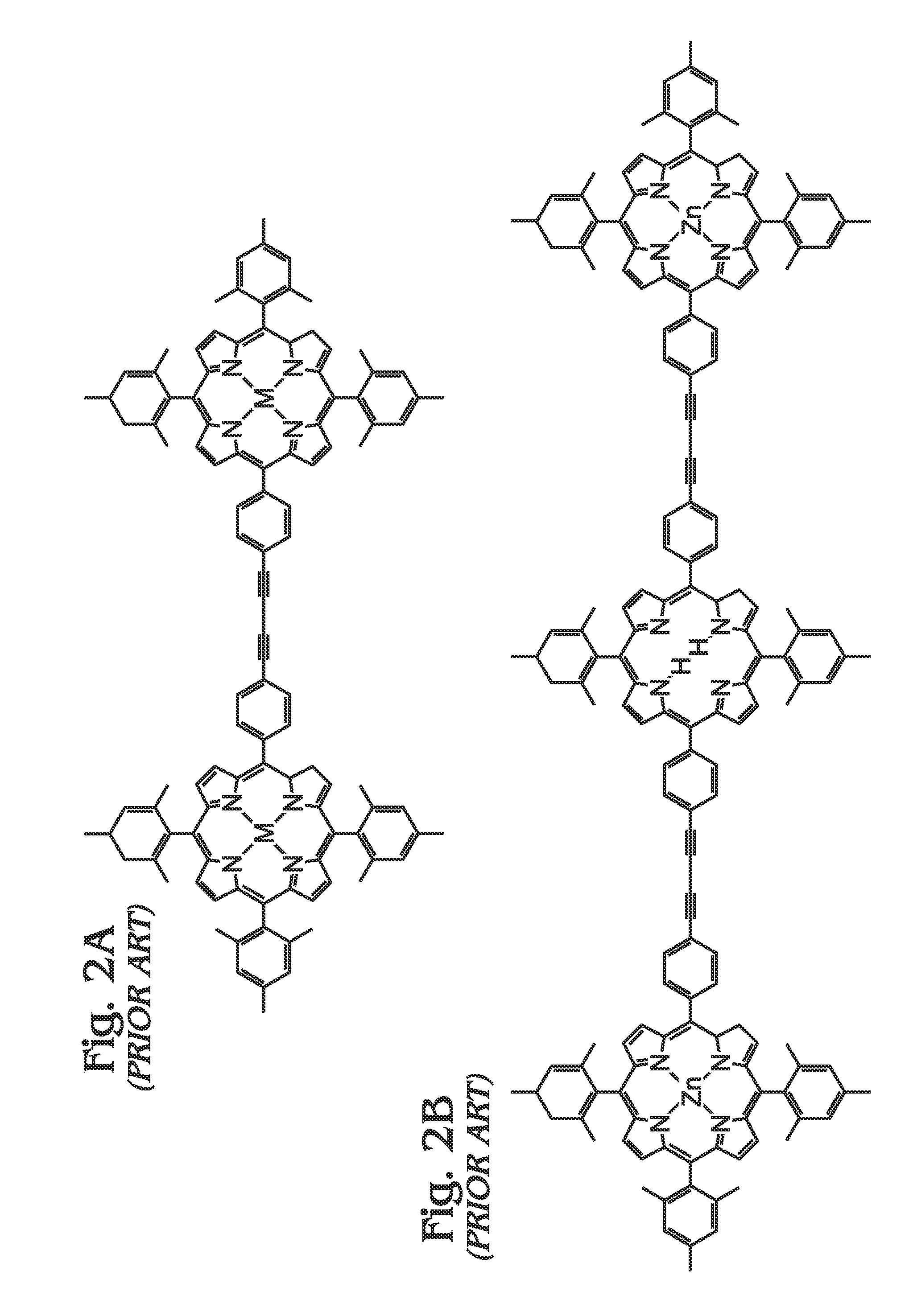
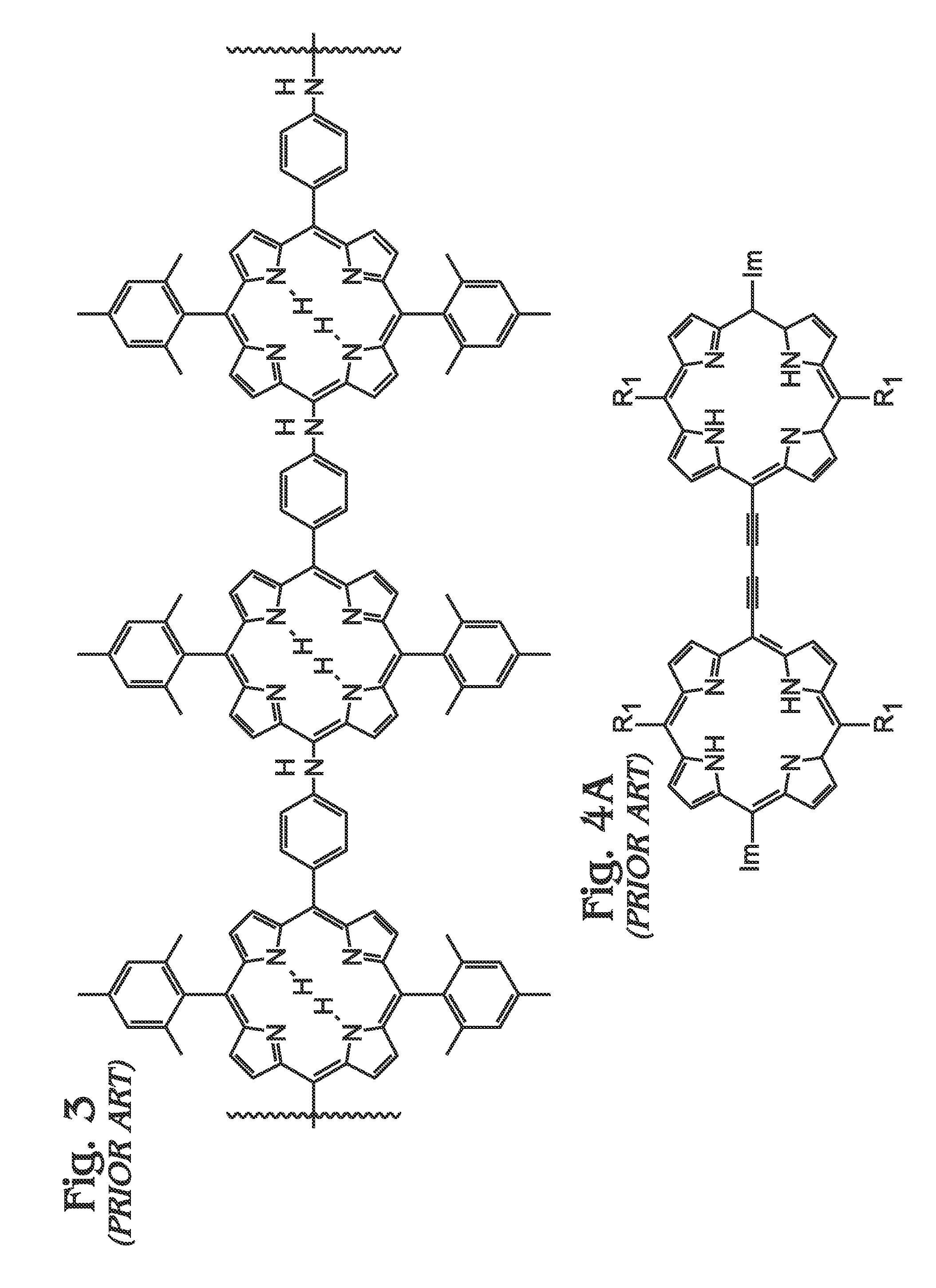
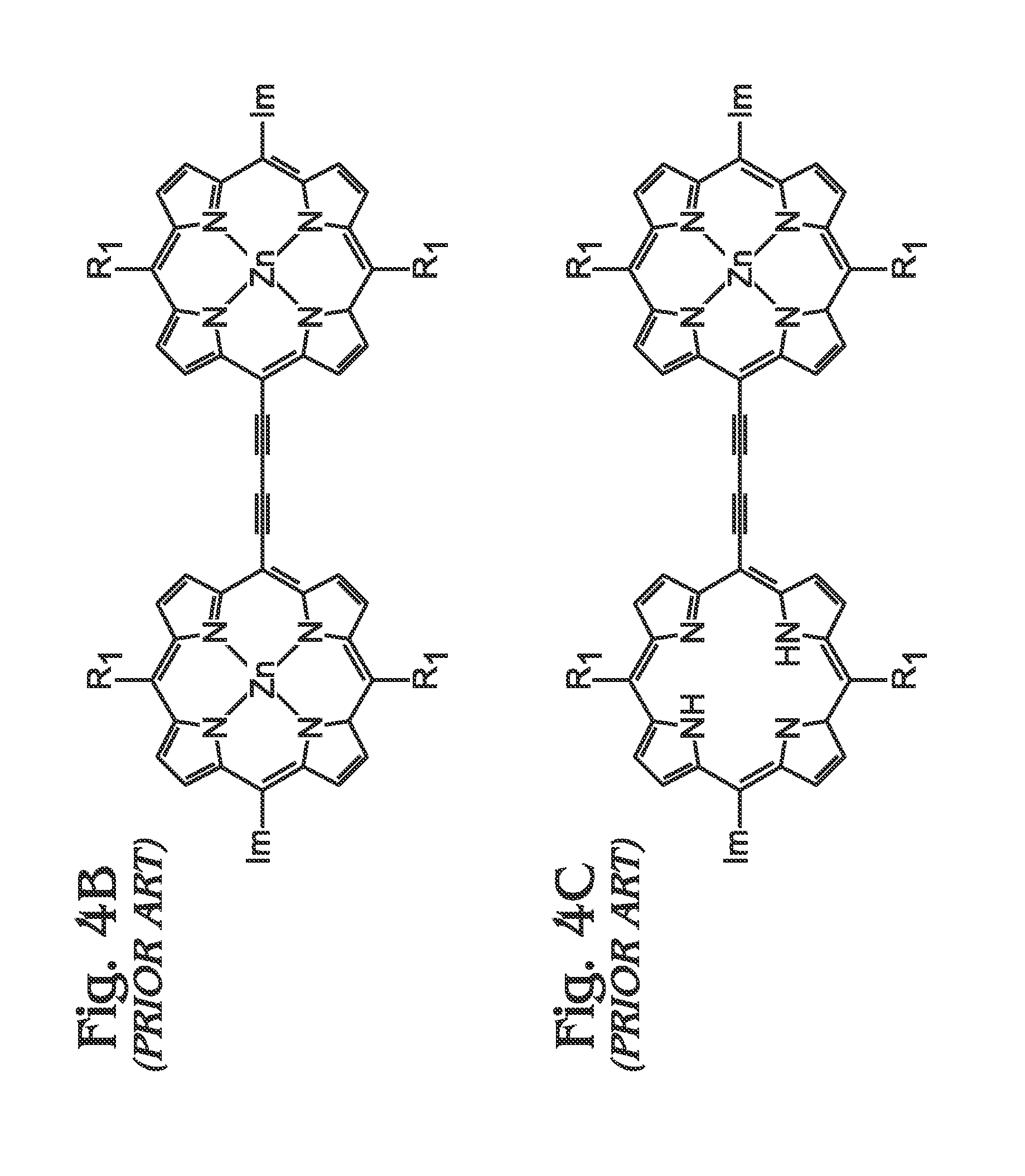
1. Field of the Invention This invention generally relates to light harvesting porphyrin polymer materials and, more particularly, to a metal (M)-meso-tetraphenylporphyrin polymer (M-poly-meso-TPP) and associated synthesis method. 2. Description of the Related Art Although chlorophyll, chlorophyll derivatives, and synthetic porphyrins have diverse molecular structures, they exhibit characteristic optical properties over comparable wavelength ranges (typically λ=350 to 700 nm in many cases). Synthetic porphyrins (and corresponding metalloporphyrins) consist of a conjugated 22π electron system, 18 of which are effectively delocalized to fit the Hückel requirement for aromaticity. In addition to their structural resemblance to natural chromophores such as chlorophyll, synthetic porphyrins are attractive candidates as light-harvesting materials due to their high structural stability, robust light absorption capabilities, and synthetic accessibility, as compared to more complex, naturally occurring chromophores. Photo-excited processes involving porphyrins are facilitated by the highly delocalized n-system, which is capable of resisting major structural changes upon oxidation. Most importantly, the redox properties of porphyrins and metalloporphyrins are dramatically altered upon photo-excitation, which leads to the generation of porphyrin excited states that can be exploited in photovoltaic (PV) applications. The ability of porphyrins to efficiently harvest light over broad wavelength ranges has generated significant interest in their potential in solar energy applications over the last few decades. As a result, synthetic protocols towards the fabrication of “customized” porphyrin architectures have become well-established and have been widely adopted as conventional methods. In general, the electronic properties of porphyrins can be altered using a number of strategies including some of the following: functionalization and/or modification along the porphyrin periphery, insertion of transition metals into the macrocyclic core, and coordination of metalloporphyrins with various ligands. Although many alternatives have evolved, there exist two major fundamental synthetic strategies for constructing the core porphyrin macrocycle, which are described independently below. In the first scenario (Method A), an aromatic aldehyde (for example, benzaldehyde in the case of tetraphenylporphyrin) or mixture of chemically functionalized aromatic aldehydes is reacted with pyrrole (or chemically functionalized pyrroles) using an acid catalyzed condensation reaction performed in organic acid (acetic acid or propionic acid, for example) at elevated temperatures. Since the distribution of chemical reaction products (porphyrins) consists of a statistical mixture representing all possible combinations at different ratios, in addition to polymeric and oligomeric products, the usefulness is ordinarily limited to symmetrical tetraphenylporphyrins. In addition, the sensitivity of the starting materials to the rather harsh reaction conditions (high temperature in acidic media) necessarily places limitations on the nature of reactants that can be utilized. Assuming the target porphyrin is an unsymmetrical tetraphenylporphyrin, subsequent purification is almost always a requirement. In almost every case, the necessary purification involves chromatographic separation of a complex mixture of porphyrin materials. Although overall reaction yields using this strategy are typically only modest, starting materials are often commercially available. As used herein, chromatography refers to a purification method that involves separating/isolating individual compounds from a mixture of compounds. The most common chromatography technique is column chromatography whereby a vertical (usually glass) column is packed with a stationary phase (usually silica gel or alumina) that functions as an adsorbent material. Typically, a material or mixture of materials (dissolved in a solvent) is placed onto the top of the stationary phase in the column and is allowed to proceed down the column by addition of a mobile phase (commonly referred to as eluent). The column chromatography process may be performed by relying on gravity (or percolation) to flow the mobile phase down the column or by applying a positive pressure in a technique commonly referred to as “flash” chromatography. During the chromatography process, an equilibrium is established between the solute (which refers to the materials/compounds to be separated) adsorbed on the stationary phase and the mobile phase flowing down the column. Owing to the differences in interactions with the stationary and mobile phases, individual components of a mixture will move down the column at different rates (partitioning), thereby allowing separation of a mixture into individual components. Typically, the eluent is collected at the bottom of the column in fractions corresponding to individual materials obtained from separation of the original mixture. Overall, the effectiveness of a chromatographic separation to provide a single component in pure form is dependent upon a number of factors including choice of stationary and mobile phases (solvent polarity, for example), partitioning coefficients for components in the original mixture, quantity of mixture to be subjected to the method at one time, and dimensions of the column in which the separation is performed, among others. In the second strategy (Method B), the porphyrin macrocycle is constructed under milder reaction conditions which is facilitated by catalytic amounts of strong acid (trifluoroacetic acid (TFA), for example) or Lewis acid (boron trifluoride (BF3), for example) in organic solvents at ambient conditions or only slightly elevated temperatures, and is usually performed under inert atmosphere (nitrogen). In many cases, an appropriate oxidizing agent (p-chloranil or similar) is added following condensation to facilitate porphyrin formation from the intermediate porphyrinogen species. In general, the milder reaction conditions are better tolerated by a wider range of starting materials, although certain functional groups still require appropriate (chemical) protection. Conveniently, elaborate porphyrin materials are more accessible due to the inherent synthetic flexibility of this approach. Although aldehydes and pyrroles may still function as the fundamental synthetic building blocks, dipyrrylmethanes, which represent a reaction product of one aldehyde with two pyrrole units, can be prepared and subsequently employed as a starting material for porphyrin formation. In this way, the amount of desired porphyrin product formed can be maximized (within statistical limits) while side-reactions are suppressed, at least to an extent. In addition to the synthesis of tetraphenylporphyrins, this strategy can be extended to the preparation of mono-, di-, and triphenylporphyrins, among others. In spite of these advantages, subsequent purification of the reaction mixture still requires chromatographic methods. In addition, the preparation of the dipyrrlmethane starting materials can prove difficult and requires analogous purification methods. Nevertheless, reaction yields for porphyrins synthesized through these methods can approach 50% in some cases. Subsequent deprotection of functional groups (if used as starting material for porphyrin formation) necessitates an additional synthetic chemistry step as well as further purification, often via column chromatography. The chemistry outlined in the two preceding methods leads to the formation of an intact porphyrin macrocycle. More often than not (at this stage), the porphyrin product functions as a scaffolding upon which additional chemical functionalities are introduced through an array of available synthetic methodologies. Such modifications, which may include the introduction of additional functional groups along the porphyrin periphery and/or insertion of transition metals in the porphyrin core, necessarily require additional synthetic steps. Each subsequent modification of the porphyrin macrocycle requires chemical reaction(s) followed by a purification step, which in most cases involves column chromatography and/or recrystallization. In addition, many of these chemical reactions require highly controlled conditions (dry solvents, inert atmosphere, etc.) and/or extended reaction times (up to 48 hours or more) and are further complicated by the fact that conversion percentages (from porphyrin starting material(s) to desired porphyrin product) can vary from extremely low to moderate, while almost never furnishing a single porphyrin product. As a result of this, chromatographic separation of a mixture of porphyrin products or, at the very least, separation of desired porphyrin product from unreacted porphyrin starting material, is required. For these reasons, only modest amounts of final porphyrin material are most often obtained following a sequence of multiple synthetic steps. As previously mentioned, the motivation to synthesize more elaborate porphyrin architectures is correlated with a desired function or performance. In simple cases, straightforward modifications are performed for the purposes of providing “attachment” to other molecules, substrates and/or other porphyrins including the introduction of carboxyl, sulfonate, or phosphonate groups for adsorption onto metal oxides (dye-sensitized solar cells, DSCs) or introduction of an aldehyde group, as is the case for constructing molecular electronic devices involving porphyrins and fullerenes or carbon nanotubes through the well-known Prato reaction. Often, the desired enhancements accessible through chemical modification of porphyrins involve manipulation of light-harvesting properties and/or excited-state behaviors (electron transfer). For example, it is well-known that increasing the n-conjugation extending from the porphyrin core can lead to enhanced absorption properties which may include (1) increased absorptivity over a particular wavelength range, (2) a broadening of optical absorption over wider ranges and/or (3) shifting of absorption towards longer (or shorter) wavelengths. Not surprisingly, this can be accomplished through a number of synthetic approaches and may involve direct attachment of conjugated moieties to the porphyrin core, formation of larger, porphyrin-centered macrocyclic derivatives prepared through ring-fusion reactions, or linear (or branched) poly-porphyrin architectures through which networks (dimers, trimers, tetramers, oligomers, polymers) of porphyrin subunits are connected through various motifs. Most often, porphyrin dimers, trimers, and tetramers are constructed in a step-wise approach whereby single porphyrin units are sequentially added to the evolving tetramer. In these cases, the construction of the final porphyrin arrays involves: (1) synthesis of monomeric porphyrin subunits (or multiple subunits), (2) chemical reaction(s) to “link” two individual subunits, (3) purification of the new molecule and, finally, (4) a repeat of steps (1)-(3). Although precise control over subunit reactivity and overall configuration is possible, this approach is both challenging and time-consuming while often leading to only modest quantities of final porphyrin material. Nevertheless, elaborate porphyrin architectures can indeed been realized using these methodologies. As an alternative to dimers, trimers, etc., large polymeric porphyrin architectures (linear or branched) are accessible using conventional synthetic methodologies with the appropriate monomeric porphyrins. Although polymeric porphyrins exhibit enhanced optical and electronic properties, overall solubility tends to be a limiting factor for practical application. In addition, the fabrication of porphyrin polymers often entails specialized equipment (electropolymerization) and/or glove-boxes/Schlenk lines (organometallic chemistry), highly-controlled chemical reaction conditions (chemical reaction stoichiometry, temperature) and, in many cases, extended periods of time. Nevertheless, the formation of extended networks (or polymers) of porphyrin-based “absorbers” is a promising strategy for fabricating both strongly and broadly absorbing materials. Therein et al. described the preparation of highly-conjugated porphyrin arrays using metal-mediated cross-coupling reactions with metalloporphyrins.3The approach(es) involve porphyrin polymer formation using classical organometallic and/or oxidative coupling chemistry. The formation of both linear and branched poly-porphyrin architectures is indeed feasible using the approaches described although major drawbacks of the technology include: (1) the need to carefully synthesize and purify the appropriate porphyrin monomers, (2) critical control of reaction parameters (Schlenk line) in order for the necessary chemical reactions to be carried out in some cases, and (3) the requirement of heating under inert atmospheric conditions for extended periods of time. In addition to synthetic methodologies described above, Chen et al. reported the synthesis of porphyrin-containing polymers linked through triazole rings prepared either by click polymerization with Cu(I) catalyst or heating to afford materials exhibiting relatively high molecular weight.4Overall, polymers obtained using the Cu(I)-catalyzed pathway provided lower molecular weights due to reduced rates for molecular weight growth compared to the metal free (thermal) polymerization. Overall, the polymers exhibited solubility in common organic solvents and demonstrated thermal stability up to 350° C. Conveniently, the method offers advantages in terms of reduced by-product formation as a result of the polymerization process. Jyothish et al. provided the preparation of a series of tris(arylmethyl)ammonium-coordinated molybdenum(VI) propylidyne catalysts that enabled the efficient synthesis of ethynylene-bridged porphyrin-based arylene ethynylene polymers through alkyne metathesis.5Xiang et al. described the synthesis of two conjugated polymers consisting of alternating main chain structures of zinc porphyrin-terthiophene (P-PTT) and zinc porphyrin-oligothiophene (P-POT).6The introduction of thiophene units in the meso-aryl positions of the porphyrin provided a bathochromic shift and broadened optical absorption characteristics relative to the monomeric zinc porphyrin (PZn) in both solution and thin solid film. Furthermore, electrochemical investigation indicated appropriate energy levels for efficient charge transfer and separation at the polymer (donor) and PCBM (acceptor) interface. Finally, bulk heterojunction (BHJ) solar cells based on P-PTT and PPOT demonstrated power conversion efficiencies (PCEs) up to 0.32% and 0.18%, respectively. Shi et al. provided a donor-acceptor porphyrin-containing conjugated co-polymer (PCTTIQP) that exhibited broad absorption along the visible spectrum.7Furthermore, corresponding BHJ solar cells based upon blends of PCTTQP and PC71BM demonstrated PCE=2.5%. Natori et al. reported the synthesis of a tetraphenylporphyrin (H2TPP)-end-functionalized poly(p-phenylene) (H2TPP-PPP) exhibiting a well-controlled polymer chain structure and broad absorption across the visible region.8Finally, Wirotius et al. described the fabrication of dendrimer-like star-branched poly(ethylene oxide)s (PEOs) comprising two and three generations with Zn(II) tetraphenylporphyrin (ZnTPP) moieties located at both the core and at each branching point through a convergent approach using “click chemistry”.9 An alternative to chemical methods for the fabrication of porphyrin polymers involves direct electrochemical polymerization of the appropriate porphyrin subunits (monomers).10-11Giraudeau et al. demonstrated a convenient method for the electropolymerization of porphyrins that circumvents the difficulties involved in synthesizing a functional porphyrin monomer by directly employing a commercial zinc-β-octaethylporphyrin (ZnOEP) with 4,4′-bipyridine (bpy) in solution.12 Although porphyrin polymers have significant relevance as absorber materials in PV applications, several reports describe the gas adsorption properties and catalytic activity of functional porphyrin networks, which are briefly summarized below. Wang et al. described the synthesis of four porous polymers consisting of nickel (Ni)-porphyrin units through which Brunauer-Emmet-Teller (BET) specific surface areas up to 1711 m2/g were achieved.17Modak et al. provided a synthesis of iron (Fe)-containing, porous organic polymers (POPs) through a one-pot, bottom-up approach.18Overall, the Fe-POPs exhibited both high BET surface area with large micropores and demonstrated excellent CO2capture. Finally, Shultz et al. demonstrated the synthesis of a POP containing free-base porphyrin subunits through condensation of a bis(phthalic acid)porphyrin with tetra(4-aminophenyl)methane.19Subsequent metallation provided microporous materials incorporating either Fe or manganese (Mn) porphyrins that demonstrated catalytic activity in both olefin epoxidation and alkane hydroxylation. In spite of the novel technologies described herein, it would be advantageous to develop methodologies for (poly)porphyrin-based “absorber” materials which meet the following criteria:
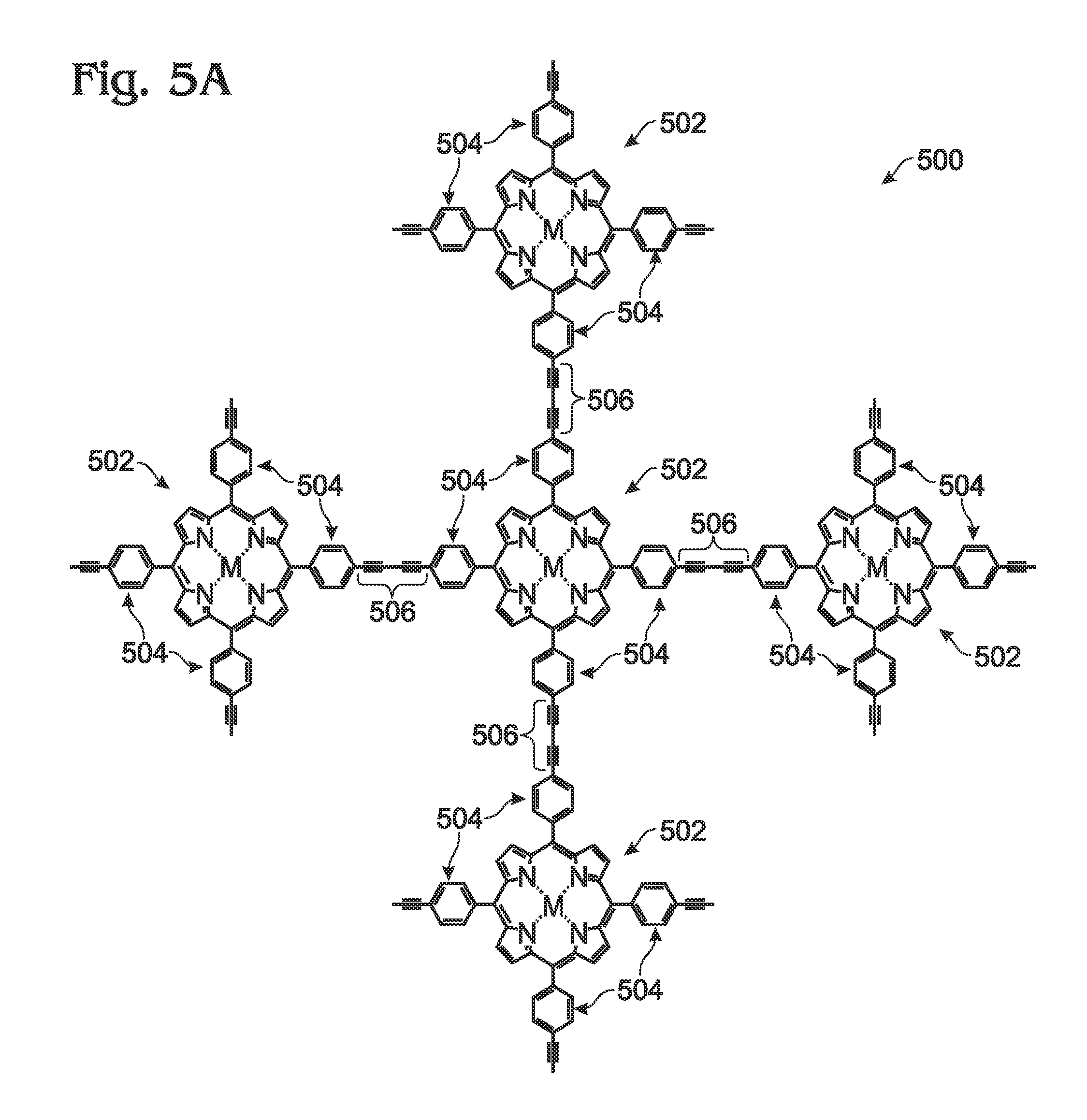
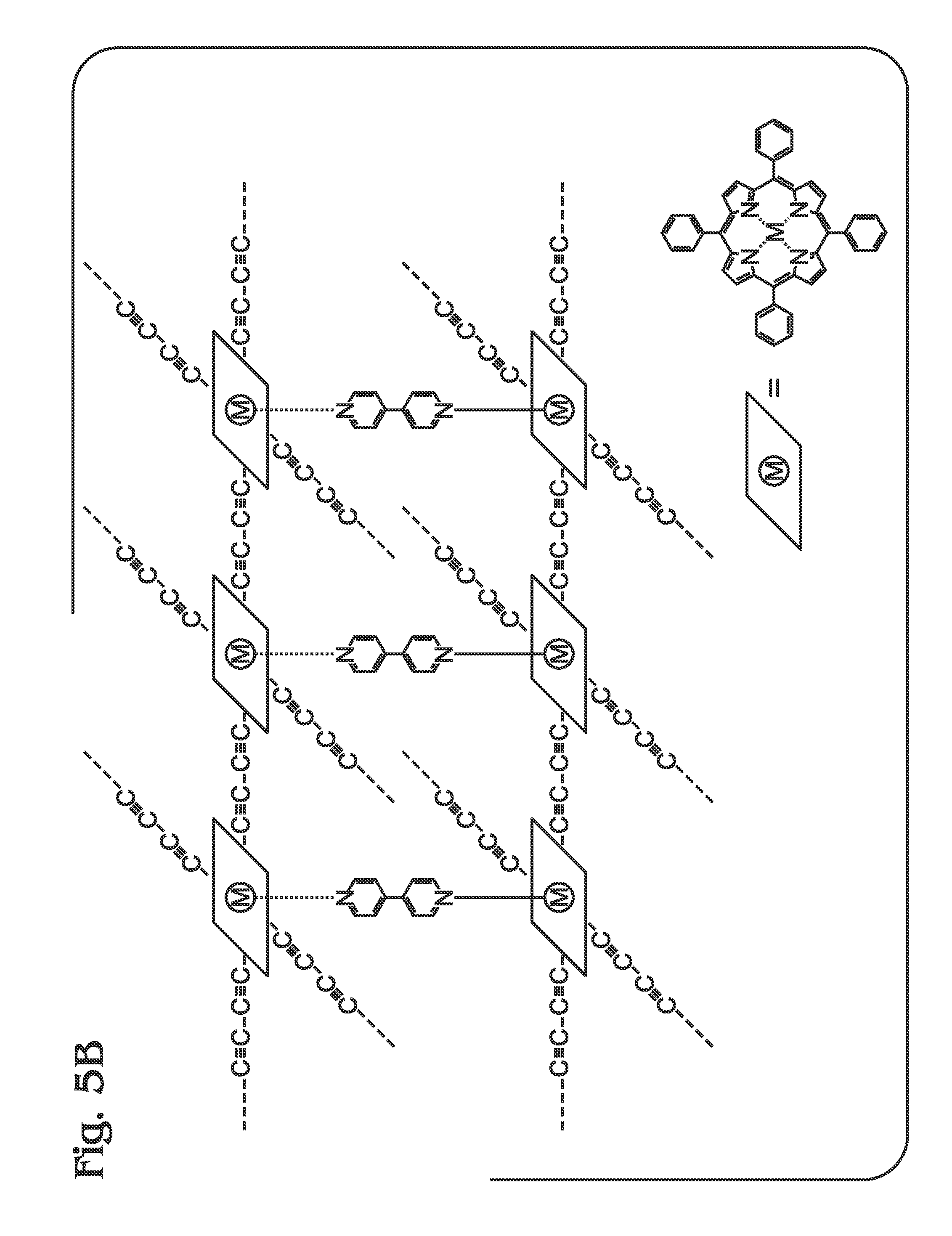
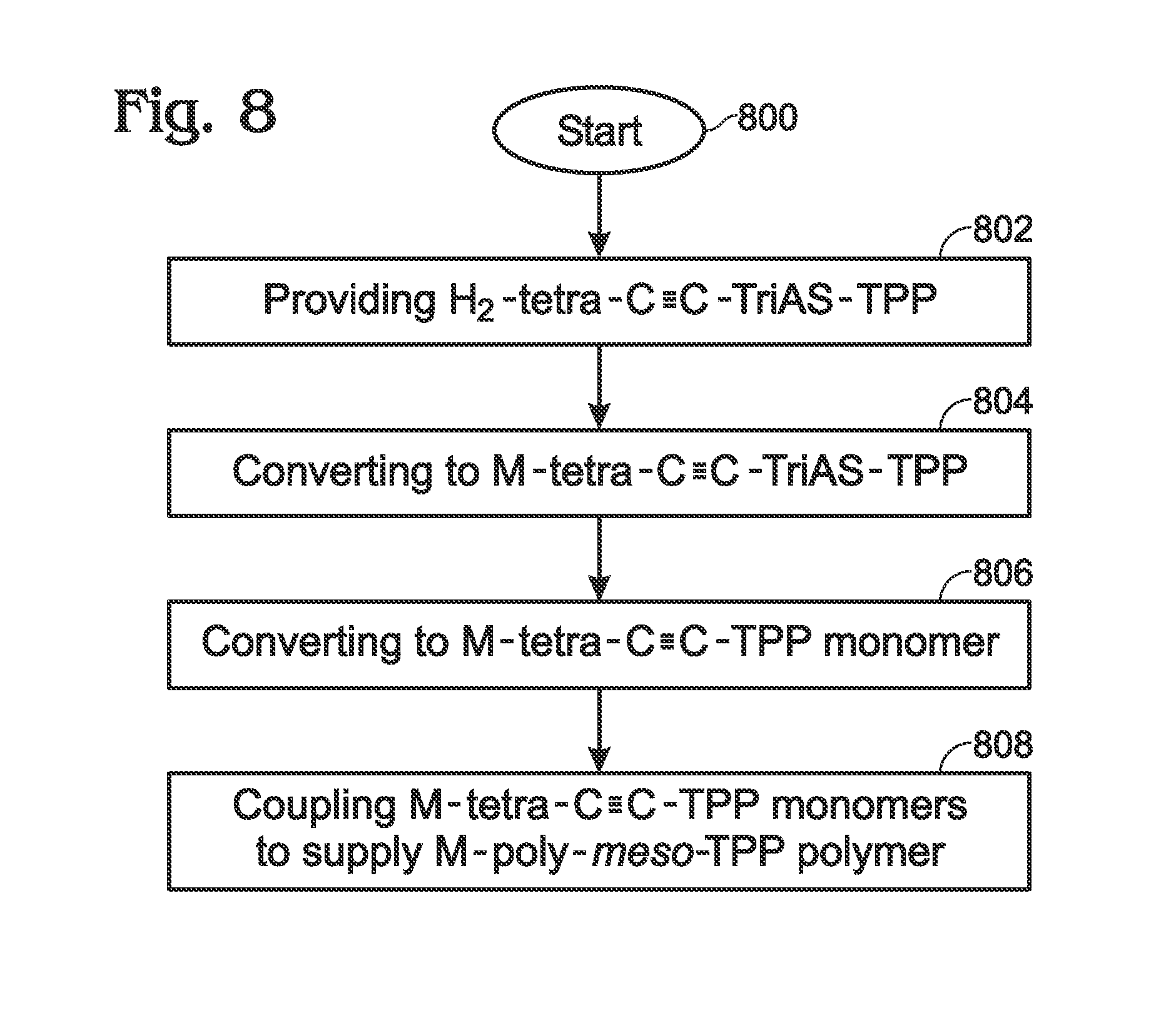
Described herein is a method for the synthesis of metalloporphyrin polymer materials, e.g., zinc porphyrin polymers, from metal (e.g. zinc) porphyrin (monomeric) subunits using a convenient methodology. The porphyrin polymer exhibits enhanced optical absorption characteristics relative to the individual porphyrin monomers and may function as a light-harvesting (“absorber”) material. The synthetic process consists of just a few steps, all of which proceed without the need for purification methods beyond straightforward filtration and, optionally, solvent evaporation following chemical reaction. Although the synthetic step affording the lowest chemical conversion yield involves formation of the porphyrin macrocycle from aromatic aldehyde and pyrrole starting materials (reactants), the subsequent chemical reactions proceed with nearly 100% conversion yield of porphyrin starting material to synthesized porphyrin product. Furthermore, all chemical manipulations can be performed without the need for environmental control. Finally, the time required for any one of the chemical reaction steps to proceed is typically around 1 hour or less. Accordingly, the synthesis of a porphyrin polymer has been successfully demonstrated using the parameters described in detail herein. Subsequent optical characterization has confirmed the enhancement in optical absorption properties for the metalloporphyrin polymer. Accordingly, a method is provided for synthesizing a metal (M) meso-tetraphenylporphyrin polymer. The method begins with the provision of a free-base (H2)-meso-tetra-4-(trialkylsilyl)ethynylphenylporphyrin (H2-tetra-C≡C-TriAS-TPP) including a trialkylsilyl (TriAS) moiety attached to an ethynyl termini. In response to a reaction with a metal (M)-containing material, the H2-tetra-C≡C-TriAS-TPP is converted to a metal (M)-tetra-4-(trialkylsilyl)ethynylphenylporphyrin (M-tetra-C≡C-TriAS-TPP). Then, the M-tetra-C≡C-TriAS-TPP is converted to a M-tetra-4-ethynylphenylporphyrin (M-tetra-C≡C-TPP) monomer by removing the TriAS moiety from the ethynyl termini. Finally, a plurality of M-tetra-C≡C-TPP monomers are coupled to supply a metal (M)-meso-tetraphenylporphyrin polymer (M-poly-meso-TPP), whereby meso-phenyl groups of adjacent M-tetra-C≡C-TPP monomers in the M-poly-meso-TPP are connected through a butadiyne linking moiety. In one aspect, the metal is zinc. Additional details of the above-described method and a metal (M) meso-tetraphenylporphyrin polymer are provided below. To a solution of 4-[(Trimethylsilyl)ethynyl]benzaldehyde in propionic acid at reflux was added dropwise with vigorous stirring an equimolar amount of pyrrole. Following heating at ˜140° C. for an additional 45 minutes, the reaction mixture was cooled, collected by filtration through a sintered glass funnel, and subsequently washed sequentially with cold MeOH and acetone. To a solution of H2-tetra-C≡C-TMS-TPP in CHCl3/MeOH (2:1) was added a stoichiometric excess of zinc acetate. The reaction was heated at reflux for 1 hour, cooled, and the solvent removed by rotary evaporation under reduced pressure. The porphyrin was suspended in cold methanol, stirred, and collected by filtration through a sintered glass funnel. Preferably, an alternative to evaporation of solvent under reduced pressure following the reaction, the Zn-tetra-C≡C-TMS-TPP product can be directly precipitated in methanol and collected by filtration as described above. Quantitative deprotection of the trimethylsilyl (TMS) protecting groups of Zn-tetra-C≡C-TMS-TPP was accomplished by introducing a stoichiometric excess of TBAF at room temperature to a solution/suspension of Zn-tetra-C≡C-TMS-TPP in THF, followed by stirring for 1 hour. Following removal of solvent by rotary evaporation under reduced pressure, the porphyrin product was suspended in cold methanol, stirred, and collected by filtration through a sintered glass funnel to afford Zn-tetra-C≡C-TPP. Preferably, as an alternative to evaporation of solvent under reduced pressure following the reaction, the Zn-tetra-C≡C-TPP product can be directly precipitated in methanol and collected by filtration as described above. A polymer comprising Zn-tetra-C≡C-TPP monomers was prepared using a copper-mediated reaction in pyridine/methanol at 25° C.→60° C. To a solution of Zn-tetra-C≡C-TPP in methanol/pyridine at 25° C. was added excess copper (II) acetate with vigorous stirring. The reaction mixture was heated and maintained at 60° C. for 1 hour, cooled, and collected by filtration using a sintered glass funnel. The polymer was washed sequentially with MeOH, water, and acetone, then collected by filtration. Since the coordination of zinc porphyrins with ligands such as pyridine can often lead to optical and/or photophysical behaviors that differ from the pristine porphyrins, absorption spectra were obtained for monomeric Zn-tetra-C≡C-TPP in mixtures of DMF and pyridine. Since the absorption spectra for Zn-tetra-C≡C-TPP showed a bathochromic shift of less than 5 nm for both the Soret and Q-bands, the spectrum for the zinc porphyrin polymer presented in Solubility (and Processability): In addition to the synthetic methodologies described above for the synthesis of M-poly-meso-TPP, strategies for enhancing the solubility may include the following: 1. Incomplete deprotection of TMS protecting groups during synthesis of the Zn-tetra-C≡C-TPP monomer from Zn-tetra-C≡C-TMS-TPP. Since polymerization of the porphyrin monomer proceeds through the terminal ethyne (alkyne) groups, retention of some fraction of the TAS groups (or TMS groups in this example) imparts solubility to the porphyrin polymer as well as functions to decrease molecular weight of the as-formed polymer. At the same time, employing alternative TAS groups (triethylsilyl, tripropylsilyl, triisopropylsilyl, for example) provides a higher degree of solubility than TMS (trimethylsilyl) when used in this fashion. In all cases, incomplete deprotection can be achieved by employing a reduced stoichiometric quantity of reagent used to affect removal of the TMS (or TAS) groups. 2. Modulation of extent for chemical coupling (polymerization). The extent (or degree) of polymerization can be controlled by the ratio of catalyst (to monomer) used during the synthesis of M-poly-meso-TPP from Zn-tetra-C≡C-TPP (monomer). For example, decreased quantities of the Cu(II) catalyst may lead to lower molecular weight materials which exhibit better solubility. 3. Introduction of moieties possessing a single polymerizable terminus (alkyne) into the polymerization reaction. These chemical species may be functionalized with organic solubilizing groups (such as alkyl groups) to impart enhanced solubility in conventional solvents while the single polymerizable terminus serves as a polymer chain terminating group that modulates the molecular weight of the porphyrin polymer. 4. Employing 3,4 dialkyl functionalized pyrroles such as 3,4-dimethylpyrrole or 3,4-diethylpyrrole instead of pristine pyrrole in the first synthetic step. Conveniently, the modification of the zinc porphyrin polymer can be accomplished via introduction of a bis-amine as ligand (for example, 1,4-diazabicyclo[2.2.2]octane (DABCO or Bipy which effectively coordinates two metalloporphyrin subunits in adjacent polymer chains, thereby restricting chain propagation to two dimensions. Finally, although the porphyrin polymer described herein refers to a metalloporphyrin containing zinc, the processes are amenable to monomeric metalloporphyrins containing other metals or mixtures of monomeric metalloporphyrins containing various metals. Step 802 provides a free-base (H2)-meso-tetra-4-(trialkylsilyl)ethynylphenylporphyrin (H2-tetra-C≡C-TriAS-TPP) including a trialkylsilyl (TriAS) moiety attached to an ethynyl termini. As is well understood, TMS (trimethyl)silyl is a member of the broader class of TAS (trialkyl)silyl species. TASs perform the same function as employed herein, which is as a chemical protecting group for terminal alkynes. Thus, although the term TMS has been used in this document as proof of concept, the function of TMS is considered to be equivalent to all TASs. Due to the appreciable chemical reactivity of terminal alkyne (ethyne) groups, appropriate chemical “protection” is employed to prevent the occurrence of undesired side reactions including acid catalyzed pathways and thermal polymerization, among other possibilities. In general, trialkylsilanes are employed due to (1) robust tolerance towards a diversity of chemical reagents and reaction conditions and (2) ease of removal in high conversion yield in a subsequent chemical reaction step. Although the identity of the alkyl group in the trialkylsilane is insignificant, a trimethylsilyl moiety was employed in the example. In response to a reaction with a metal (M)-containing material, Step 804 converts the H2-tetra-C≡C-TriAS-TPP to a metal (M)-tetra-4-(trialkylsilyl)ethynylphenylporphyrin (M-tetra-C≡C-TriAS-TPP). In one example, the metal (M) is zinc (Zn). Step 806 converts the M-tetra-C≡C— TriAS-TPP to an M-tetra-4-ethynylphenylporphyrin (M-tetra-C≡C-TPP) monomer by removing the trialkylsilyl (TriAS) moiety from the ethynyl termini. Step 808 couples a plurality of M-tetra-C≡C-TPP monomers to supply a metal (M)-meso-tetraphenylporphyrin polymer (M-poly-meso-TPP), whereby meso-phenyl groups of adjacent M-tetra-C≡C-TPP monomers in the M-poly-meso-TPP are connected through a butadiyne linking moiety. In one aspect, coupling the plurality of M-tetra-C≡C-TPP monomers to supply the M-poly-meso-TPP includes synthesizing the M-poly-meso-TPP from M-tetra-C≡C-TPP monomers in the presence of a Cu(II)-containing material and an amine (pyridine, for example). In one aspect, the Cu(II) containing material is copper(II) acetate. In another aspect, the amine is pyridine, a chemically functionalized pyridine, quinoline, or a chemically functionalized quinoline. In another aspect, the amine used during the polymerization of M-tetra-C≡C-TPP monomers remains coordinated to M-poly-meso-TPP following reaction and isolation. In another aspect, Step 808 synthesizes the M-poly-meso-TPP from M-tetra-C≡C-TPP monomers in the presence of a bisamine-containing material. For example, the bisamine-containing material may coordinate with an M moiety of adjacent M-poly-meso-TPP polymer chains, as shown in In one aspect, providing the H2-tetra-C≡C-TriAS-TPP in Step 802 includes reacting a first material, such as pyrrole or an alkyl group functionalized pyrrole, with a 4-[(trialkylsilyl)ethynyl]benzaldehyde. For example, the 4-[(trialkylsilyl)ethynyl]benzaldehyde material may be 4-[(trimethylsilyl)ethynyl]benzaldehyde. In another aspect, all the above-mentioned steps are performed using a solvent. Explicitly, Step 802 may synthesize the H2-tetra-C≡C-TriAS-TPP using chemical reactions performed in a solvent. Step 804 may synthesize the M-tetra-C≡C-TriAS-TPP using chemical reactions performed in a solvent. Step 806 may synthesize the M-tetra-C≡C-TPP using chemical reactions performed in a solvent, and Step 808 may synthesize the M-poly-meso-TPP using chemical reactions performed in a solvent. Further, all the above-mentioned steps may be performed in an ambient environment, without the requirement of a vacuum atmosphere, inert gases, or other environmental controls. Explicitly, Step 802 may synthesize the H2-tetra-C≡C-TriAS-TPP in an ambient environment. Step 804 may synthesize the M-tetra-C≡C-TriAS-TPP in an ambient environment. Step 806 may synthesize the M-tetra-C≡C-TPP monomer in an ambient environment, and Step 808 may synthesize the M-poly-meso-TPP in an ambient environment. In another aspect, all the above-mentioned steps may be performed without the requirement of chromatographic purification. Explicitly, Step 802 may sequester H2-tetra-C≡C-TriAS-TPP from solvents and reaction components in response to filtering performed without using chromatographic purification. Step 804 may sequester M-tetra-C≡C-TriAS-TPP from solvents and reaction components in response to filtering performed without using chromatographic purification. Step 806 may sequester M-tetra-C≡C-TPP from solvents and reaction components in response to filtering performed without using chromatographic purification. Step 808 may sequester M-poly-meso-TPP from solvents and reaction components in response to filtering performed without using chromatographic purification. An M-poly-meso-TPP and associated synthesis method have been provided. Examples of particular metals, reagents, and process variables have been presented to illustrate the invention. However, the invention is not limited to merely these examples. Other variations and embodiments of the invention will occur to those skilled in the art. A method is provided for synthesizing a metal (M) meso-tetraphenylporphyrin polymer. The method begins with the provision of a free-base (H2)-meso-tetra-4-(trialkylsilyl)ethynylphenylporphyrin (H2-tetra-CC-TriAS-TPP) including a trialkylsilyl (TriAS) moiety attached to an ethynyl termini. In response to a reaction with a metal (M)-containing material, the H2-tetra-CC-TriAS-TPP is converted to a metal (M)-tetra-4-(trialkylsilyl)ethynylphenylporphyrin (M-tetra-CC-TriAS-TPP). Then, the M-tetra-CC-TriAS-TPP is converted to a M-tetra-4-ethynylphenylporphyrin (M-tetra-CC-TPP) monomer by removing the trialkylsilyl (TriAS) moiety from the ethynyl termini. Finally, a plurality of M-tetra-CC-TPP monomers are coupled together to supply a metal (M)-meso-tetraphenylporphyrin polymer (M-poly-meso-TPP), whereby meso-phenyl groups of adjacent M-tetra-CC-TPP monomers in the M-poly-meso-TPP are connected through a butadiyne linking moiety. In one aspect, the metal is zinc. 1. A method for synthesizing a metal (M) meso-tetraphenylporphyrin polymer, the method comprising:
providing a free-base (H2)-meso-tetra-4-(trialkylsilyl)ethynylphenylporphyrin (H2-tetra-C≡C-TriAS-TPP) including a trialkylsilyl (TriAS) moiety attached to an ethynyl termini; in response to a reaction with a metal (M)-containing material, converting the H2-tetra-C≡C-TriAS-TPP to a metal (M)-tetra-4-(trialkylsilyl)ethynylphenylporphyrin (M-tetra-C≡C-TriAS-TPP); converting the M-tetra-C≡C-TriAS-TPP to a M-tetra-4-ethynylphenylporphyrin (M-tetra-C≡C-TPP) monomer by removing the trialkylsilyl (TriAS) moiety from the ethynyl termini; and, coupling a plurality of M-tetra-C≡C-TPP monomers to supply a metal (M)-meso-tetraphenylporphyrin polymer (M-poly-meso-TPP), whereby meso-phenyl groups of adjacent M-tetra-C≡C-TPP monomers in the M-poly-meso-TPP are connected through a butadiyne linking moiety. 2. The method of 3. The method of 4. The method of 5. The method of wherein converting the H2-tetra-C≡C-TriAS-TPP to the M-tetra-C≡C-TriAS-TPP includes synthesizing the M-tetra-C≡C-TriAS-TPP using chemical reactions performed in a solvent; wherein converting the M-tetra-C≡C-TriAS-TPP to the M-tetra-C≡C-TPP monomer includes synthesizing the M-tetra-C≡C-TPP monomer using chemical reactions performed in a solvent; and, wherein coupling the plurality of M-tetra-C≡C-TPP monomers to supply the M-poly-meso-TPP includes synthesizing the M-poly-meso-TPP using chemical reactions performed in a solvent. 6. The method of wherein converting the H2-tetra-C≡C-TriAS-TPP to the M-tetra-C≡C-TriAS-TPP includes synthesizing the M-tetra-C≡C-TriAS-TPP in an ambient environment; wherein converting the M-tetra-C≡C-TriAS-TPP to the M-tetra-C≡C-TPP monomer includes synthesizing the M-tetra-C≡C-TPP monomer in an ambient environment; and, wherein coupling the plurality of M-tetra-C≡C-TPP monomers to supply the M-poly-meso-TPP includes synthesizing the M-poly-meso-TPP in an ambient environment. 7. The method of wherein converting the H2-tetra-C≡C-TriAS-TPP to the M-tetra-C≡C-TriAS-TPP includes sequestering M-tetra-C≡C-TriAS-TPP from solvents and reaction components in response to filtering performed without using chromatographic purification; wherein converting the M-tetra-C≡C-TriAS-TPP to the M-tetra-C≡C-TPP monomer includes sequestering M-tetra-C≡C-TPP monomer from solvents and reaction components in response to filtering performed without using chromatographic purification; and, wherein coupling the plurality of M-tetra-C≡C-TPP monomers to supply the M-poly-meso-TPP includes sequestering M-poly-meso-TPP from solvents and reaction components in response to filtering performed without using chromatographic purification. 8. The method of 9. The method of 10. The method of 11. A metal (M) meso-tetraphenylporphyrin polymer (M-poly-meso-TPP) comprising:
a plurality of M-tetra-C≡C-TPP monomers with meso-phenyl groups; and, butadiyne linking moieties connecting the meso-phenyl groups of adjacent M-tetra-C≡C-TPP monomers. 12. The M-poly-meso-TPP of 13. The M-poly-meso-TPP of BACKGROUND OF THE INVENTION
SUMMARY OF THE INVENTION
BRIEF DESCRIPTION OF THE DRAWINGS
DETAILED DESCRIPTION
Experimental (Synthetic) Details
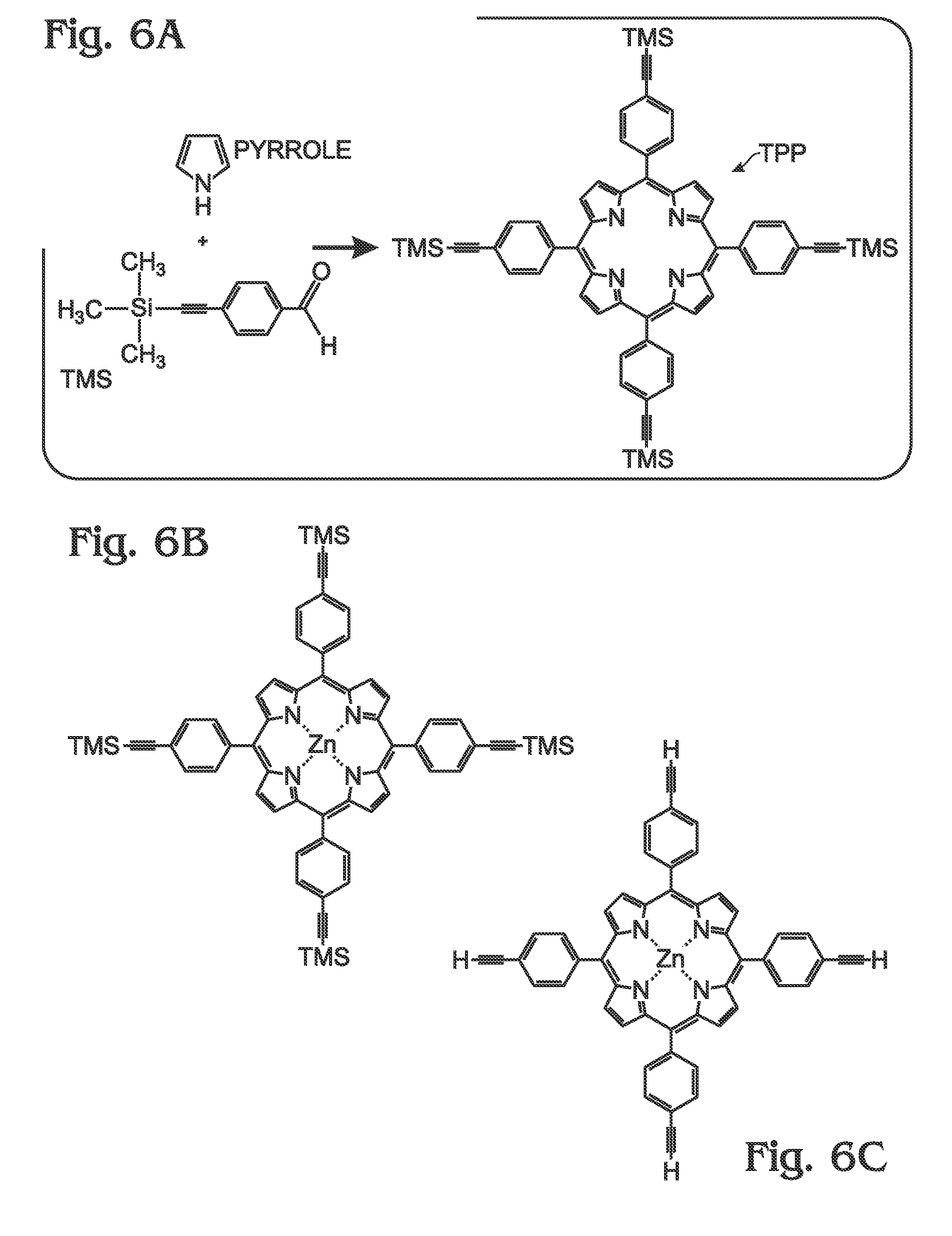
H2-tetra-4-(trimethylsilyl)ethynylphenylporphyrin (H2-tetra-C≡C-TMS-TPP) (Step 1, see FIG. 6A)
Zinc-tetra-4-(trimethylsilyl)ethynylphenylporphyrin (Zn-tetra-C≡C-TMS-TPP) (Step 2, FIG. 6B):
Zinc-tetra-4-ethynylphenylporphyrin (Zn-tetra-C≡C-TPP) (Step 3, FIG. 6C)
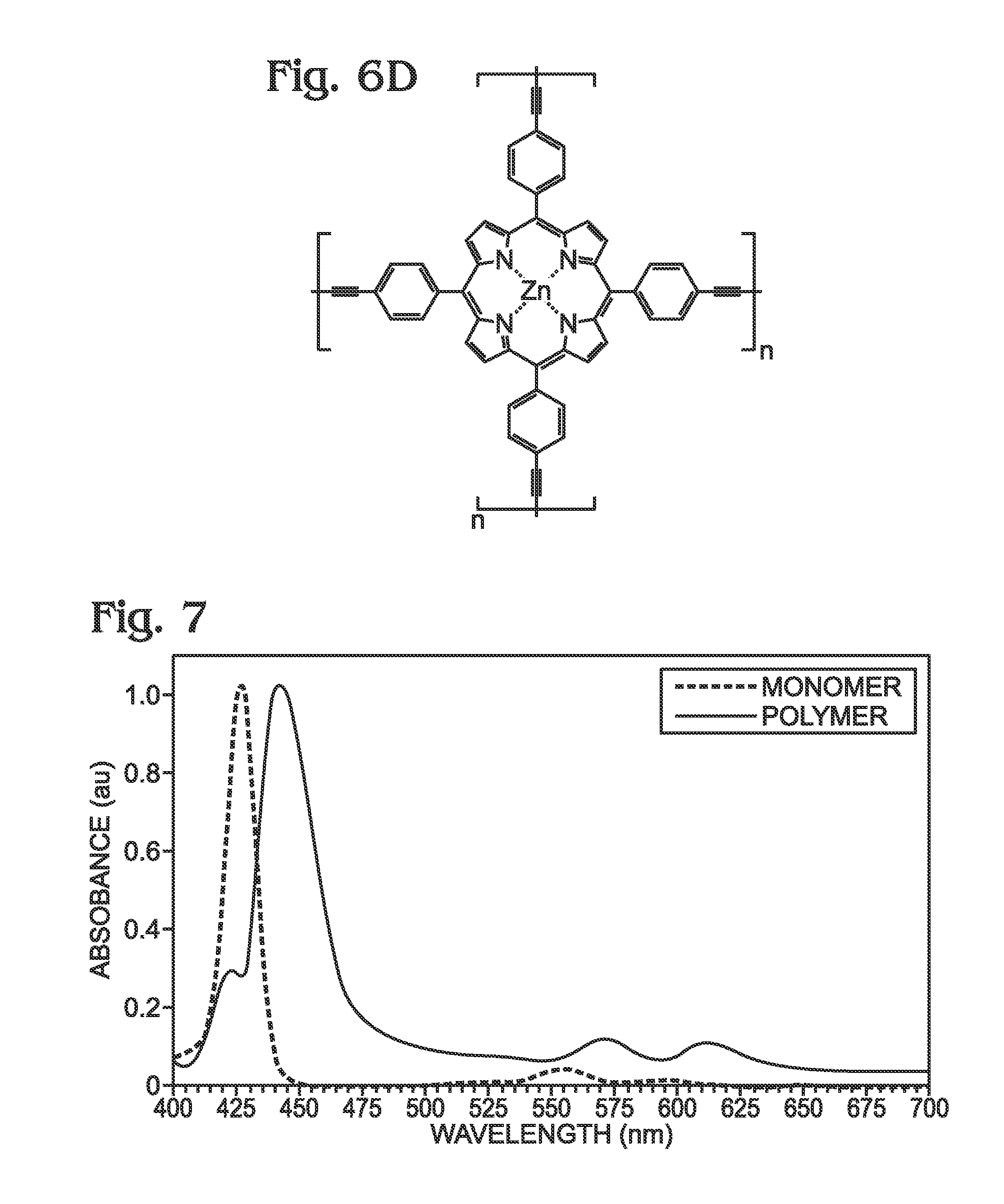
Zinc Porphyrin Polymer (M-poly-meso-TPP) (Step 4, FIG. 6D)
Optical Absorption Spectrum:
Modification of Zinc Porphyrin Polymer Morphology: