MODIFIED PEGYLATED RECOMBINANT HUMAN INTERFERON Α 2B AND METHOD FOR PREPARING SAME
本发明属于生物制药领域,涉及可修饰氨基酸在干扰素中的定点插入及修饰方法,以及获得的新型改良型长效干扰素,以及非天然氨基酸介导的不同单PEG修饰产物,特定位点不同分子量的PEG修饰,以及多位点定点PEG的修饰产物。 (1)干扰素 干扰素是细胞在诱生剂作用的条件下产生的一种蛋白质,具有抑制病毒复制,抑制细胞分裂,及免疫调节等功能。人干扰素α 2b分子量约为19kD,由165个氨基酸组成,其序列如SEQ ID NO:1所示: 根据文献报道,对个别密码子进行优化,以提高其在大肠杆菌中的表达量,所用编码人干扰素α 2b的编码序列如SEQ ID NO:2所示: 干扰素是商品化较早的生物制品,自从1986年美国FDA批准Roch公司的干扰素α 2a及Schering公司的干扰素α 2b上市,目前干扰素已经成为世界上基因工程药物的重要成员。干扰素在我国也是第一个投放市场的基因药物,已经获得了广泛的使用。其在我国慢性乙型肝炎等治疗方面取得的重要效益和经济利益,将使干扰素成为未来抗病毒,抗癌症最广泛应用的药物之一。 作为基因药物,干扰素半衰期短而治疗周期长,需要患者频繁的注射给药,大大降低了患者的依从性以及干扰素的应用价值。因此,改善干扰素药代动力学的研究正在世界范围内广泛进行。 (2)聚乙二醇修饰 共价交联聚乙二醇是增加生物分子水溶性,调节免疫原性,延长其半衰期的常用方法。利用聚乙二醇修饰干扰素,延长其半衰期,改善其生物学特性,已经是得到应用的成功技术。此前已经有美国先灵葆雅公司生产的佩乐能,及罗氏公司生产的派罗欣作为PEG化干扰素通过FDA认证进入了医药市场。但是,此两种药物所使用的PEG连接方法都可以和干扰素上多个位点发生反应,因此会产生多聚体和同
分异构体。作为细胞因子类药物,干扰素发挥生物学效应需要和相应的受体结合,而非理想位点的不可控PEG修饰会明显阻碍干扰素与其受体分子结合,从而会大大减少PEG化干扰素的生物学活性;而多PEG化及单PEG同分异构体的产生,又对工业生产的后期处理带来高成本的影响,且由于同分异构体的分离的复杂性,在工业化生产中分离同分异构体的步骤往往被省略。已上市的两种PEG化干扰素都为多位点单PEG修饰的同分异构体混合物,相比于未修饰干扰素,其生物学活性大大下降。 针对此问题,国内外已经发表了多篇关于单PEG化修饰的干扰素方法。半胱氨酸的定点突变修饰PEG是单修饰的常用方法,其利用半胱氨酸上特有的巯基作为活性基团,通过特异性地和活性PEG反应进行修饰(Mary S.Rosendahl et al,Bioconjugate Chem.2005;Stacie J.Bell et al,Bioconjugate Chem.2008),此种方法虽然可以一定程度上解决单PEG修饰的异构体问题,但对氨基酸序列中已有半胱氨酸的蛋白质而言,引入多余的半胱氨酸未必可以达到理想的效果,可能会造成蛋白质的多聚化,二硫键的错配等严重问题;Sibu Balan等对干扰素进行定点的二硫键修饰,得到了单PEG修饰的干扰素(Sibu Balan et al,Bioconjugate Chem.2007),但二硫键往往参与形成蛋白质的活性口袋,并对蛋白质形成正确的折叠起到重要作用,也因此成为针对二硫键的定点修饰技术的严重限制;另外,也有通过改变PH以达到定点N端的PEG修饰方法(Amartya Basu et al,Bioconjugate Chem.2006),以及利用Dock-and-lock融合蛋白技术
修饰PEG(Chien-Hsing Chang et al,Bioconjugate Chem.2009)等,但都无法回避位点的局限性以及修饰条件的复杂性问题。 利用叠氮和炔的Click反应在生物分子体系中进行修饰是近年来研究的热门领域。Natalie W.Nairn等利用甲硫氨酸缺陷型菌在干扰素β中引入含有叠氮基团的非天然氨基酸,然后利用有铜催化的Click反应进行单位点的PEG修饰(Natalie W.Nairn et al,Bioconjugate Chem.2012)。但是此种方法仅能针对蛋白质中甲硫氨酸位点引入设计好的非天然氨基酸,如果蛋白质中有多个甲硫氨酸位点,要达到单PEG修饰的目的,则需要定点突变其余位点的甲硫氨酸,如此以来,无论在位点选择的灵活性上,及操作的复杂性上,都有着很大的限制。 (3)遗传密码扩展技术 近年来遗传密码扩展技术发展迅速,利用琥珀终止密码子(TAG)为有义编码子,通过引入相应的正交tRNA及氨酰tRNA合成酶,最终可以将设计好的非天然氨基酸引入蛋白质中。到目前为止,这一技术已经将几十种非天然氨基酸成功地定点表达在活细胞的蛋白质当中,涉及的非天然氨基酸含有炔基,叠氮等,利用这些生物体中本来不存在的特殊基团,就可以特异性的对蛋白质进行定点修饰。 面对现有技术中干扰素修饰不均匀,分离纯化工艺复杂等问题,在干扰素的PEG化研制中,迫切需要能够任意位点特异性修饰功能基团的方法。而用此方法得到的聚乙二醇化干扰素,也有望能够得到更优良的性质。
发明内容 发明人经过对现有技术的思考,将非天然氨基酸技术应用到干扰素药物的修饰上,利用古甲烷球菌的tRNA(tRNAPyl)和吡咯赖氨酰-tRNA合成酶(tRNAPyl/PylRS)的蛋白质翻译系统使含有叠氮基团非天然氨基酸定点掺入到蛋白中,从而得到定点突变的干扰素。通过在不同位点引入含有叠氮基团修饰把手的非天然氨基酸,进而对干扰素实现了任意位点的定点修饰。通过活性筛选及性质测定,得到了若干性质优良的PEG化干扰素产品。随后发明人进一步应用非天然氨基酸的修饰方法,对干扰素进行了多点的定点修饰,得到了不错性质的双点修饰PEG的干扰素产品。 相比于现有的修饰方法及上市的PEG化干扰素产品,本发明使用的修饰方法及得到的改良型PEG化干扰素产品主要优势体现在如下几点: 1.可以在干扰素的任意位点引入非天然氨基酸,从而创造可以仅对该位点进行特异性修饰的原料PEG化干扰素前体; 2.利用非天然氨基酸上特有的叠氮活性基团,以及环辛炔介导的无铜Click反应,可以对干扰素实现特异性,高效,无毒害,简单易行的修饰反应。 3.任意位点的PEG修饰可以带来更好的修饰效果,修饰在非活性位点,有可能实现药效减少最小化和药代性能最优化的修饰目的;修饰在抗原暴露区,可以实现免疫原性降低的效果;修饰在易被蛋白酶水解区,可以实现有效避被免体内蛋白酶降解目的; 4.得到的PEG化干扰素修饰均一化,批次之间质量控制容易; 5.得到的PEG化干扰素在保证药代动力学性质优良的前提下,拥有更高的保留活性。 具体的,在本发明的一个具体的实施方案中,提供了不同位
点引入含有叠氮基团非天然氨基酸的人干扰素,主要通过三个步骤:(1)选择可能合适的干扰素突变位点,(2)构建含有在选定的具有TAG突变的编码人干扰素基因的载体,(3)获得pSUPAR-YAV-tRNAPyl/PylRS质粒,将步骤(2)与(3)在合适的宿主菌中共表达,且在培养基中添入需要的非天然氨基酸,经过后续的纯化步骤,获得在不同位点含有叠氮基团非天然氨基酸(NAEK)的干扰素突变体,命名为XY(NAEK)-IFN,X指代原有位置氨基酸种类,Y指代序列位置,例如H34NAEK-IFN或H34-IFN为非天然氨基酸NAEK替代34位组氨酸所得到的干扰素突变体。 该突变系统的原理在于:突变型的tRNAPyl/PylRS满足下列关系:(1):突变型的tRNAPyl不能利用宿主细胞的赖氨酰tRNA酶,只能被突变型的PylRS酰化;(2):突变型的PylRS只能酰化tRNAPyl,不能酰化其它tRNA,因此,突变型tRNAPyl和PylRS之间的关系是正交性的,即突变型的PylRS只能酰化突变型tRNAPyl,同时突变型的tRNAPyl只能被突变型的PylRS酰化,也就是说同一质粒中的突变型的tRNAPyl和PylRS是绝对的相互专一的。这种正交性的酶并且是只有这种酶可以把非天然氨基酸酰化到这种正交的tRNA上,并且只能酰化这种tRNA,而不能酰化其它的tRNA。获得的正交赖氨酰tRNA合酶/tRNA系统,使非20种常见氨基酸的Lys-azido(也可称为:NAEK)与琥珀密码子相对应,从而将非天然氨基酸定点引入到目的蛋白(例如人干扰素)中。 在本发明的一个具体实施方案中,本发明选取了OrigamiB(DE3)表达体系,此种感受态细胞通过基因改造,缺失蛋白还原途径的两个关键酶,从而稳定了蛋白质的二硫键,使菌体内蛋白更易形成天然构象,增强蛋白可溶性。 在本发明的一个具体的实施方案中,通过对未经PEG修饰的干扰素突变体进行活性筛选,从而得到插入非天然氨基酸对干扰素活性影响比较小的干扰素突变体,并将其做为修饰的候补产物。 在本发明的一个具体的实施方案中,将适合修饰的候补产物
与修饰剂进行无铜催化的Click反应,修饰剂为以环辛炔为连接物的聚乙二醇,使得分子量5k,10k,20k,40k的直线型或分枝型PEG通过干扰素上的非天然氨基酸对干扰素实行定点修饰,经简单离子交换色谱纯化即可获得特定位点定点PEG化的干扰素。 在本发明的一个具体的实施方案中,通过对候补产物进行PEG化,并对PEG化后的产物进行体外活性的筛选,得到了相比于上市PEG化干扰素而言,体外活性更高并且修饰均一的PEG化干扰素及其若干候补修饰产物。 在本发明的另一个实施方案中,通过上述筛选得到的对干扰素活性影响较小的突变位点,发明人选取两个对活性影响最小的突变位点,进行双点的PEG修饰,得到更有利于保留活性及提高药代动力学参数指标的干扰素修饰产物。 更为具体地,本发明提供了 1.定点突变的人干扰素,其在特定位点的1个氨基酸被突变为非天然氨基酸,所述非天然氨基酸为:如式(I) 所示的NAEK,即Lys-azido 所述特定位点选自:示于SEQ ID NO:1的第P4位,H7位,S8位,K31位,R33位,H34位,E41位,E51位,A74位,E78位,G102位,T106位,E107位,L110位,M111位,E113位,L128位,K133位,P137位,E159位或其他对活性影响较小的位点. 2.编码突变的干扰素的核酸分子,所述核酸分子与编码SEQ ID NO:1的核酸分子SEQ ID NO:2的区别在于,其中编码SEQ ID NO:1的第P4位,H7位,S8位,K31位,R33位,H34位,E41位,E51位,A74位,E78位,G102位,T106位,E107位,L110位,M111位,E113位,L128位,K133位,P137位,E159位或其他对活性影响较小的位点的一个氨基酸的密码子被突变为TAG。 3.经过修饰的定点突变的干扰素,其连接方式如下式(II)所示,其中,其中,R1为突变前目的蛋白(例如SEQ ID NO:1)
所示序列的第1至第N-1位氨基酸残基, R2为突变前目的蛋白(例如SEQ ID NO:1)所示序列的第N+1位至C末端的氨基酸残基,R3为相同或不同分子量PEG,当被取代的氨基酸不止一个时,其上连接的PEG分子量可以相同也可以不同。 4.核酸载体,其可操作地连接有项目2的核酸分子。 5.宿主细胞,其中含有项目4的核酸载体,还含有表达甲烷球菌的tRNA(tRNAPyl)和吡咯赖氨酰-tRNA合成酶(tRNAPyl)的质粒。 6.一种含有非天然氨基酸的干扰素定点突变体的获得方法,包括步骤: (1)选择步骤:在干扰素的氨基酸序列中选择期望突变的一个或多个特定氨基酸位点; (2)表达载体的构建:构建野生型人干扰素的基因载体,然后用定点突变的方法将编码对应于(1)中选择的位点的干扰素的氨基酸的密码子用基因工程方法突变为密码子TAG,得到含有突变序列的表达载体; (3)获得pSUPAR-YAV-tRNAPyl/PylRS质粒:从保藏日为2011年6月14日、保藏号为CGMCC No:4951的大肠埃希氏菌
pSUPAR-YAV-tRNAPyl/PylRS中获取质粒pSUPAR-YAV-tRNAPyl/PylRS质粒; (4)表达:将(2)得到的含有突变序列的表达载体与(3)的pSUPAR-YAV-tRNAPyl/PylRS质粒共同转染相同的宿主细胞,将转染成功后的宿主细胞在含有NAEK的培养基中培养,并在合适的条件下诱导表达; (5)对表达产物进行分离提取及纯化,得到特定位点含有叠氮基团的干扰素突变体 7.筛选得到适合进行PEG修饰的干扰素突变体位点,包括对项目6得到的突变体进行光学表面等离子共振实验(SPR)及Daudi细胞的抗肿瘤增值活性的测定,得到适合修饰的干扰素突变体候补产物。 8.进一步筛选得到适合进行PEG修饰的干扰素突变体,主要步骤包括如下: (1)对项目7得到的候补产物进行10kD大小PEG的修饰,得到PEG化后的干扰素候补产物 (2)对(1)中得到的产物进行活性测定的筛选,其中包括Daud i细胞的抗肿瘤增值活性,光学表面等离子共振实验(SPR),脑心肌炎病毒(EMCV)与A549细胞的抑制细胞病变效应,圆二色谱鉴定二级结构,以及体内稳定性的评价,得到保留活性较强的PEG化干扰素突变体 (3)重新表达并纯化(2)中筛选得到的具有该位点非天然氨基酸的干扰素突变体 (4)对(3)中得到的突变体进行5kD,10kD,20kD,40kD的PEG连接 (5)对(4)中得到的产物再次进行活性评价,以得到在该修饰位点更利于修饰的PEG分子大小 9.双点修饰PEG干扰素的获得及活性测定.主要步骤如下: (1)选择项目8中筛选得到的两个较利于进行PEG修饰的人
干扰素突变位点 (2)表达并纯化两个不同的选定位置被取代为含叠氮基团的非天然氨基酸的干扰素突变体 (3)将(2)中的突变体与PEG反应进行偶联,通过分离纯化,得到特定位点双点PEG修饰的干扰素突变体 (4)对(3)中得到的突变体进行活性评价 10.制备定点PEG化的干扰素的方法,包括在合适的条件下将项目1、3或项目9获得的定点突变的干扰素与适量的活性PEG进行Click反应,得到用聚乙二醇定点修饰的突变的干扰素。 11.定点改良的干扰素,其在项目1或3的定点突变的干扰素的非天然氨基酸位置定点引入PEG,以及在项目9中双点突变位置引入PEG 12.组合物,其中含有有效量的项目1、3、11中任一项的干扰素,项目3的经过修饰的定点突变的干扰素或者项目9的定点改良的干扰素。 13.药物组合物,其中含有有效量的项目1、3或11中任一项的干扰素,项目3的经过修饰的定点突变的干扰素、项目9的定点改良的干扰素,以及药学上可以接受的赋形剂。 14.项目1、3或11中任一项的干扰素,项目3的经过修饰的定点突变的干扰素、项目9的定点改良的干扰素、项目12或13的组合物在制备长效,稳定性干扰素,用于抗病毒,治疗多种恶性肿瘤,免疫调节的药物中的用途。 本发明所述表达突变的tRNAPyl/PylRS的质粒可来源于从保藏日为2013年4月8日、保藏号为CGMCC No:7432的大肠埃希氏菌pSUPAR-YAV-tRNAPyl/PylRS中获取的pSUPAR-YAV-tRNAPyl/PylRS质粒。 本发明还提供了一种微生物(例如大肠杆菌),其含有本发明的表达突变的tRNAPyl/PylRS的质粒。示例性地,本发明的微生物为保藏号为CGMCC No:7432的微生物。
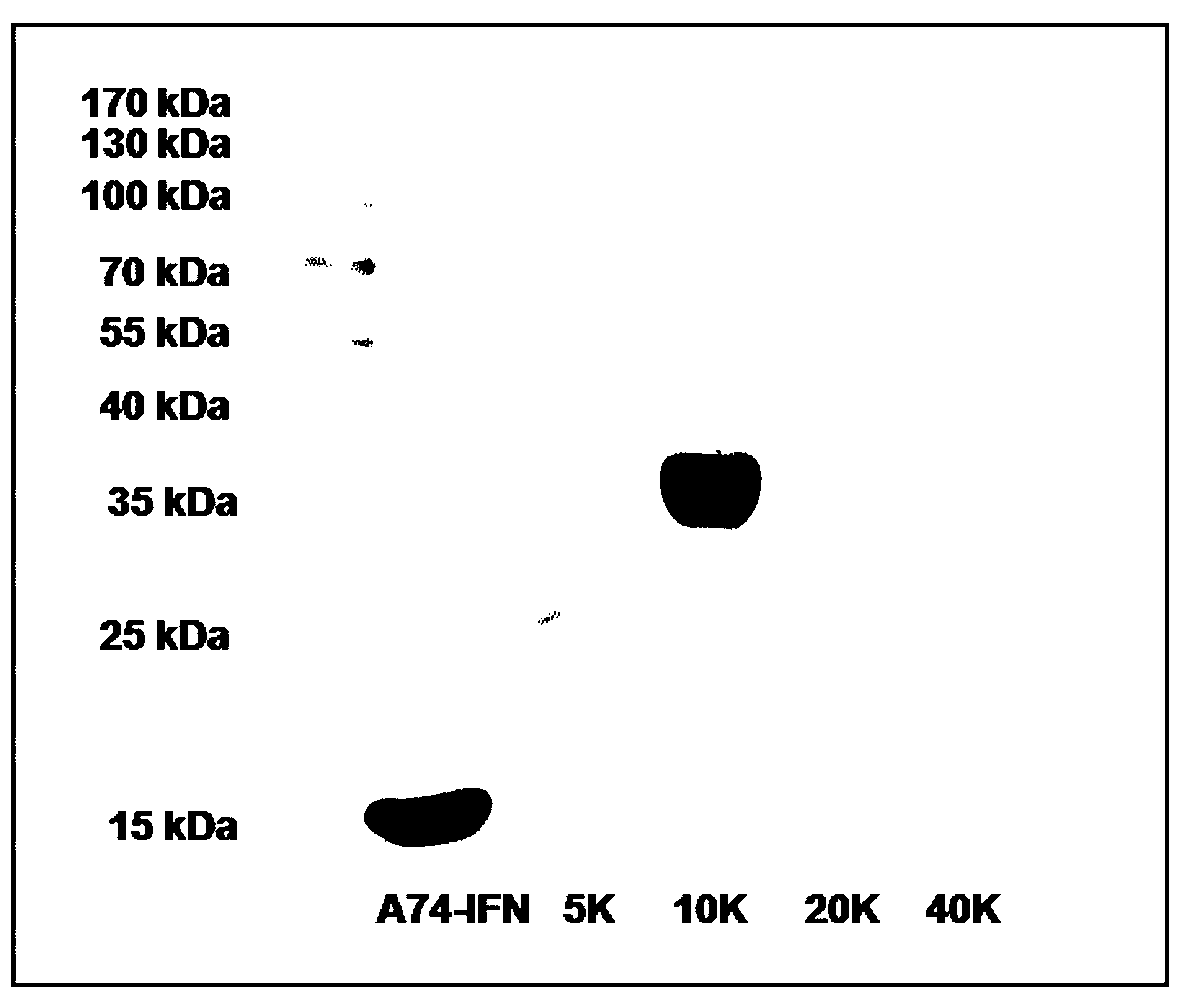
图1:经PEG修饰的突变型型人干扰素α 2b 第1道为重组表达的A74突变为为NAEK的人干扰素α 2b 第2道至第5道依次分别为经5k Da、10k Da、15k Da、20k DaPEG分子修饰的人干扰素α 2b 图2:突变以及进行PEG化的干扰素与野生型干扰素的质谱分析结果。 图2-A:选定位置(E41)被取代的干扰素的质谱结果 图2-B:PEG化的定点修饰的干扰素质谱结果 图3:干扰素突变体(这里指特定位点取代了非天然氨基酸,而未进行PEG修饰的产物)的体外活性柱状图汇总. 图3-A:各突变体体外抗病毒活性(a:自己表达、未突变的干扰素;b:阴性对照;c:市场购买干扰素)、其中P4表示干扰素氨基酸第四位由非天然氨基酸替换原有的脯氨酸(P)的干扰素,其他标注以此类推 图3-B:各干扰素突变体与干扰素受体2的平衡解离常数 图4:各干扰素突变体PEG化后的体外活性柱状图汇总 图4-A:体外抗病毒活性(PEG-Intron为市售PEG化干扰素,此作为对照) 图4-B:体外抗肿瘤活性 图4-C:与受体2的平衡解离常数 图5:干扰素各突变体PEG化后的物理性质测定 图5-A:WT-IFN,H7、H34、A74、E107、E113-10K-IFN圆二色谱测定结果,WT-IFN表示野生型的IFN,H7、H34、A74、E107、E113-10K表示在该特定位点发生突变,并修饰10kDa PEG的干扰素 图5-B:热稳定性的检测结果,其中WT-IFN表示野生型的IFN,H34-5K-IFN、H34-10K-IFN、H34-20K-IFN、H34-40K-IFN分别表示发生了H34突变,并经分子量为5K、10K、20K、40K Da的PEG修
饰的干扰素 图5-C:SD大鼠体内的药代动力学结果。其中WT-IFN表示野生型的IFN,H34-10K、A74-10K、E107-10K分别表示发生了H34、A74、E107突变,并经分子量为10K Da的PEG修饰的干扰素 图6:干扰素H34和E107位点双位点突变并修饰PEG的实验结果 图6-A:H34/E107双位点修饰物与单位点修饰的SDS-PAGE结果。第一道为未修饰PEG的H34&E107-IFN,第二道为H34-IFN与5K-PEG反应的产物,第三道为H34&E107-IFN与5K-PEG反应的产物,第四道为H34-IFN与10K-PEG反应的产物;第五道为未修饰PEG的H34&E107-IFN,第六道为E107-IFN与20K-PEG反应的产物,第三道为H34&E107-IFN与20K-PEG反应的产物,第四道为E107-IFN与40K-PEG反应的产物 图6-B:PEG化的H34&E107双位点修饰圆二色谱的鉴定结果,其中WT-IFN表示野生型的IFN,H34-5K、H34-10K、H34-20K、H34-40K分别表示发生了H34突变,并经分子量为5K、10K、20K、40K Da的PEG修饰的干扰素,H34&E107-20K表示发生了H34和E107双突变,并经分子量为20KDa的PEG修饰的干扰素。 图7:H34和E107双位点突变修饰PEG的干扰素的体外活性 图7-A:不同样品体外抗病毒活性。H34&E107-5K表示发生了H34和E107双突变,并经分子量为5KDa的PEG修饰的干扰素;H34&E107-20K表示发生了H34和E107双突变,并经分子量为20KKDa的PEG修饰的干扰素;对照组为WT、即野生型干扰素、H34位点及E107单位点修饰5、10、20、40kDa PEG的干扰素产品、PEG-Intron为市售PEG化干扰素 图7-B:不同样品体外抗病毒活性。各标注同图7-A 图7-C:不同样品受体结合活性。各标注同图7-A 图8不同的构建体对胰酶的耐受实验结果。其中WT-IFN表示野生型的IFN,H34-5K-IFN、H34-10K-IFN分别表示发生了H34
突变,并经分子量为5K、10K Da的PEG修饰的干扰素,H34&E107-5K-IFN表示发生了H34和E107双突变,并经分子量为5KDa的PEG修饰的干扰素。 图9对34位进行了不同PEG分子量修饰的药代动力学的体内试验结果。 图9-A:各样品静脉注射曲线,WT表示野生型的IFN,H34-5K、H34-10K、H34-20K、H34-40K分别表示发生了H34突变,并经分子量为5K、10K、20K、40K Da的PEG修饰的干扰素,H34&E107-5K、H34&E107-20K表示发生了H34和E107双突变,并经分子量为5K、20KDa的PEG修饰的干扰素 图9-B:各样品皮下注射曲线、各样品标注同9-A 为了更好地理解本发明,发明人用实施例对具体试验进行阐述和说明,其中所述实施例仅用于说明,并不限定本发明的保护范围。任何与本发明等价的变体或者实施方案都包括在本发明中。 实施例1:包含定点突变的人干扰素的基因载体的构建 (1)天然人干扰素质粒辅助质粒的获得 经全基因合成,获得干扰素的基因(SEQ ID NO:2)。然后将其连接在带有6*His标签的pET-21a(+)表达载体中,获得天然干扰素的表达质粒(pET21a(+)-IFN(WT);从中国微生物菌种保藏管理委员会普通微生物中心(菌种保藏地址:北京市朝阳区北辰西路1号院,中国科学院微生物研究所,保藏日为2013年4月8日、保藏号为CGMCC No:7432的分类命名为大肠埃希氏菌(Escherichia coli))的含有质粒pSUPAR-YAV-tRNA/PylRS的大肠埃希氏菌pSUPAR-YAV-tRNA/PylRS中获取质粒pSUPAR-YAV-tRNA/PylRS(以下简称该质粒为辅助质粒),该质粒可以表达特异识别非天然氨基酸Lys-azido的tRNA和tRNA合成酶。
(2)定点突变位点的选择及突变载体的构建 根据干扰素的晶体结构,干扰素与其受体的结合位点,不同干扰素亚型的保守序列以及氨基酸暴露范围,并综合考虑抗原表位,酶解位点等信息[Ramaswamy Radhakrishnan,Leigh J Walter,Zinc mediated dimer of human interferon-a2b revealed by X-ray crystallography,Structure 1996,Vol 4No 12;Christoph Thomas et al,Structural linkage between ligand discrimination and receptor activation by type I interferons,Cell.2011 August 19;146(4):621–632.],本发明的发明人选取了几个适合位点进行修饰,其主要基于以下因素:1.氨基酸暴露于蛋白表面以方便偶联;2.屏蔽免疫原区;3.屏蔽蛋白酶解区。 通过更详细的文献资料查阅[Mary S.Rosendahl et al,Bioconjugate Chem.2005,16,200-207;Stacie J.Bell et al,Bioconjugate Chem.2008,19,299–305],以及对比已上市的PEG化干扰素的高活性PEG修饰异构体,发明人选取第P4位,H7位,S8位,K31位,R33位,H34位,E41位,E51位,A74位,E78位,G102位,T106位,E107位,L110位,M111位,E113位,L128位,K133位,P137位,E159位为特定位点进行点突变,以此突变型干扰素为原料并对其进行定点修饰。 针对上述位点,发明人设计能够使编码所述氨基酸的密码子突变为琥珀密码子的引物, (3)突变载体构建 发明人针对人干扰素第P4位,H7位,S8位,K31位,H34位,E51位,A74位,G102位,T106位,E107位,M111位,Y129位,K133位,K134位,P137位,E159这几个位点,分别设计能够使编码所述氨基酸的密码子突变为琥珀密码子的引物,然后利用定点突变试剂盒(Lightning Site-Directed Mutagenesis Kits,Catalog#210518),按说明书操作以上述步骤(2)中获得的野生型干扰素表达载体pET21a(+)-IFN(WT)为模板将干扰素相
应位点的密码子突变为琥珀终止密码子,得到突变型干扰素的质粒。 (4)定点突变的干扰素表达株的构建 将步骤(1)得到的带有氯霉素抗性的辅助质粒和步骤(3)得到的带有氨苄青霉素抗性的表达质粒同时转化大肠杆菌OrigamiB(DE3),经氯霉素和氨苄青霉素双抗性平板筛选出共转化的阳性菌株,即同时转化有两个质粒的菌株。 实施例2:定点突变的干扰素的表达、纯化、PEG化以及鉴定 本发明中构建pSUPAR-YAV-tRNA/PylRS质粒与表达源自古甲烷球菌的tRNA(tRNAPyl)和吡咯赖氨酰-tRNA合成酶(tRNAPyl)的质粒共表达后,在宿主菌中,从原理上看,利用这套蛋白质翻译系统能够使非天然氨基酸NAEK掺入到蛋白中,从而造成干扰素的定点突变。 发明人对NAEK的掺入可能性进行了实验,并对定点的PEG化进行了鉴定。 1:突变干扰素的NAEK掺入表达及纯化 (1)将实施例1的步骤4获得的表达株在含有34ug/ml氯霉素和100ug/ml氨苄青霉素的2*YT培养基中37℃培养12-16小时后,再经二级扩增至菌液OD值到0.6-1.0时,加入NAEK至终浓度1mM,37℃继续扩增30分钟,加入IPTG至终浓度0.5mM,阿拉伯糖至终浓度0.1%,24℃诱导表达12小时后收集菌体。 (2)将收集的菌体用Ni-NTA-Bind-Buffer平衡重悬,经超高压匀质破碎机1200bar,2循环破碎后,高速离心去除细胞碎片,经过Ni-NTA金属螯合亲和层析,用Ni-NTA-Wash-Buffer充分洗涤,最后用Ni-NTA-Elute-Buffer洗脱,得到初步纯化的干扰素样品,纯度约为90%. 2.突变体的聚乙二醇定点偶联及纯化 无铜催化的Click反应需借助环辛炔的环张力作用实现,将
修饰物与DIBO(有环辛炔结构的化合物)偶联,从而与含有叠氮的基团进行无铜催化Click反应(Mbua,N.E et al,ChemBioChem.2011,12,1912-1921) 通过无铜催化Click反应偶联PEG,反应体系如下: 经实施例2中的步骤1所制备的NAEK-IFN 1μg/μl DIBO-PEG 2mM 反应条件:4℃,垂直混悬2小时。 经过上述反应条件能够在1小时内将大约50%的干扰素定点PEG化,反应后的复合物经过除盐,再经过离子交换(Source 15S,20mM乙酸钠PH=4.5,0-250mM NaCl梯度),可以获得>95%纯度的PEG定点修饰蛋白。结果验证不同分子量PEG都可成功修饰到干扰素上,并实现纯化(见图1) 3:非天然氨基酸的定点插入,及定点PEG化的鉴定(见图2) 将突变及进行PEG化的干扰素与野生型干扰素进行SDS-PAGE电泳,经考马斯亮蓝染色及脱色后,进行生物质谱分析。由质谱结果可见,选定位置天然氨基酸已被非天然氨基酸NAEK所替代(图2-A),而替代位置已定点修饰上PEG(图2-B) 实施例3:适合进行修饰的干扰素突变体获得 干扰素序列中氨基酸的改变会对干扰素的性质产生影响,通过评价单点替换非天然氨基酸对干扰素体外活性带来的影响,初步的对适合进行非天然氨基酸插入及其后修饰的干扰素突变体进行筛选及确定。 A.光学表面等离子共振实验(SPR): 干扰素α 2b通过结合细胞表面两个受体单位发挥生物学作用,我们称之为IFNAR1和IFNAR2。其中IFNAR2是主要的结合单位,以纳摩尔的高亲和力与α类干扰素结合。 光学表面等离子共振实验(SPR)通过将一个蛋白锚定在传感芯片表面,然后将待测样品流过芯片表面,若样品中有能够与
芯片表面的生物分子识别膜相互作用的分子,就会引起金膜表面折射率变化,最终导致SPR角变化,通过检测SPR角度变化,获得被分析物的浓度、亲和力、动力学常数和特异性等信息。 具体过程如下:将IFNAR2锚定在CM5芯片上,响应值230RU;以30ul/min,120s进样留过干扰素样品;解离时间120s;4M氯化镁,30ul/min,洗涤20秒;通过留过不同浓度样品,由软件拟合KD值。通过比较KD值,量化突变氨基酸以及修饰PEG对干扰素活性造成的影响。 B.抗肿瘤增值活性: 抗肿瘤活性是干扰素重要生理作用之一。干扰素诱导的2-5A合成酶-RNASE L以及PKR-eIF2系统与细胞的增殖相关,而且2-5A合成酶和RNase L酶在快速生长的细胞中已经被发现高水平的表达,表明他们在调控细胞的增长方面发挥着重要作用。 评价干扰素的体外抗肿瘤活性是对干扰素质量评价的一个有效指标。具体做法如下:将对数生长的Daud i细胞2万个每孔接种于96孔板中;37℃,5%CO2培养约1小时;加入不同梯度的干扰素及突变体、PEG修饰物,2100pg/ml-0.9pg/ml,1:3倍比稀释,以等体积空白培养基作为空白对照,以不加干扰素,仅加入缓冲液作为阴性对照;约96小时后用Celltiter-Glu法评价细胞活性,绘制剂量依赖曲线,并计算IC50。 为了得到适合在干扰素中进行PEG修饰的位点,发明人通过上述体外实验,对表达的野生型即未引入非天然氨基酸以及通过实施例2所制备的突变型干扰素进行SPR及抗Daudi肿瘤细胞的增殖实验,结果如表1所示: 表1:野生型及突变型干扰素的SPR及抗肿瘤增值活性IC50 *阴性对照(备注:根据报道,干扰素R33位点的突变会导致其体外活性大量下降,即KD值及IC50值理论上会显著上升,以此作为阴性对照,以说明实验方法及过程的正确性) 根据上述实验结果,基于以下两点考虑:(1)在未修饰PEG情况下,突变体体外活性较高的;(2)所选位点应均匀分布于干扰素序列不同位置;发明人选取P4、H7、H34、E41、E51、A74、E107、E113、P137-IFN作为插入非天然氨基酸影响较小的干扰素突变体,以及适合进行PEG化修饰的候补产物进行后续实验。
实施例4:高活性、修饰均一的PEG化干扰素获得 考虑到与上市的PEG化标准品PEG-Intron,12kDa-PEG做对比,发明人选择10kDa-DIBO-PEG对实施例3筛选获得的干扰素修饰候补产物进行修饰,并通过体外活性及其他性质的测定对修饰产物进行评价。 A.抑制细胞病变效应 作为抗病毒蛋白药物,干扰素具有广谱的抗病毒增值作用,进而保护细胞免受病毒侵害。人干扰素α 2b的抑制细胞病变效应也是广泛使用的评价干扰素体外的体外活性的重要指标之一。具体做法如下:按2*104个/孔接种A549细胞于96孔板,过夜培养(约12-16小时),加入梯度浓度的各干扰素样品刺激细胞,于第三日弃培养上清液,加入稀释好的EMCV病毒液,量足以使90%以上细胞发生细胞40小时内发生细胞病变效应,约40小时后用Celltiter-Glu法评价细胞活性,计算半数保护量,并与人干扰素α 2b标准品(PBL公司购买)进行标定,计算比活性。 B.圆二色谱 蛋白质的肽键在紫外185-240nm处有吸收,具有圆二色性。在蛋白质中,不同二级结构所表现出的圆二色谱是不同的。蛋白质总体的圆二色谱是他们所含各种立体结构组分的圆二色谱的代数加和曲线,用该范围内的圆二色谱可以研究蛋白质中各种立体结构的含量。对干扰素进行非天然氨基酸的插入及PEG的修饰有可能改变干扰素的二级结构,进而影响其活性功能,因此对修饰后的干扰素突变体进行圆二色谱的测定。具体做法如下:用PBS将待测样品统一稀释至0.3ug/ul,并于圆二色谱仪上(JASCO J-810)上在190-240nm波长处进行测定。 C.热稳定性 通常的蛋白质在高温下很容易变性,进而聚集形成沉淀丧失其功能活性。PEG的修饰会使蛋白质的溶解度增加,阻止蛋白质的
相互聚合,从而显著增加蛋白质对抗热的稳定性。另外,热稳定性的评价也是对修饰反应化学键稳定与否的有效指标。具体做法如下:将待测样品用PBS统一稀释至0.3ug/ul,取100ul,在干式恒温仪中65℃加热,于不同的时间点(5、10、15、20、25min)取样,在405nm波长处进行浑浊度的测定。 D.体内稳定性 干扰素的主要缺点之一为体内半衰期短,通过PEG的修饰,增加其分子体积大小,能够有效的减少其肾小球滤过率,屏蔽其酶解位点,延长干扰素的体内稳定性。因此测定PEG化后干扰素的体内稳定性能够很有效的评价修饰效果。具体做法如下:两只体重约300g的雄性SD大鼠为一组,静脉注射100ug/kg的待测样品,分别于0h、0.25h、0.5h、1h、2h、4h、6h、24h、48h、72h眼静脉取血入EDTA抗凝管中,12000g离心,分离血浆于-80℃保存。随后对各血样进行A549/EMCV的活性检测,以确定各时间点的保留活性,并折合为浓度,最后通过软件绘制曲线并计算药代动力学参数。 将实施例3中筛选得到的突变体进行10kD的PEG修饰,经分离纯化后,得到各位点单点PEG修饰的突变体,随后进行抗肿瘤增值,SPR,及抑制细胞病变效应的体外活性评价,结果如表2所示: 表2:PEG化干扰素突变体体外活性总结 a为各待测样品的抑制细胞病变效应保留百分比,记WT为100% *为上市PEG化干扰素对照品,购自先灵葆雅公司(备注:此上市的PEG化干扰素为12kDa-PEG非定点修饰的干扰素α 2b产品,根据文献报道,其体外抗病毒活性约保留27%) 由表2可以看到,34位点定点修饰的干扰素H34-10K-IFN相对于其余的修饰突变体,有着最高的体外保留活性,其相对于上市的PEG化干扰素PEG-Intron,其抗肿瘤活性及抑制细胞病变效应有着将近1倍的提高;而A74、E107-10K-IFN,相比于PEG-Intron,有着相似的体外活性数据,可作为候补PEG化产物 选取WT-IFN,H7、H34、A74、E107、E113-10K-IFN进行圆二色谱的测定(图3-A),结果表明各类型的PEG修饰对其二级结构没有明显影响;随后对WT-IFN、H34-5K/10K/20K/40K-IFN进行热稳定性的检测(图3-B),结果显示,相比于未修饰PEG的干扰素(WT-IFN),修饰了PEG的干扰素在热稳定上有显著的提高;SD大鼠体内的药代动力学数据(图5-C)表明,H34/A74/E107-10K-IFN相比于WT-IFN,其体内稳定性有着显著提高,其半衰期延长了5-6倍,清除率明显下降。 为了更好的平衡干扰素的体内稳定性及生物活性,发明人对修饰的PEG分子量进行了优化。具体做法为通过评价不同PEG分子修饰对干扰素受体结合能力、体外抗肿瘤活性、抑制细胞病变效应的活性影响,来选择更适合在特定位点进行修饰的PEG分子,具体结果如表3所示: 表3.PEG大小对特定位点干扰素活性影响总结
由结构可以总结出:各位点都不适合进行大分子量的PEG修饰(40K相比20K,体外活性有明显降低);20K的PEG修饰相比10K,有着相似的体外活性。在大分子量PEG修饰可能更好的提高稳定性的前提下,提示20K的PEG可能为更好的修饰剂。而H34-10K,H34-20K,A74-10K,E107-10K在较大PEG分子的修饰下,表现除了较好的体外活性,可选为候补产物。 实施例5:双点插入非天然氨基酸及PEG修饰的干扰素 通过上述实例的描述,可以看出单一位点的定点PEG修饰可以有效的提高PEG干扰素的体外活性,通过筛选得到的H34-10K-IFN可以保留干扰素40%以上的体外活性。为了尝试更理想性质的PEG化干扰素获得,发明人首次尝试了多点PEG化对干扰素活性及性质的影响。相比于单点的PEG修饰,多点的优势可能体现在如下几点:1)分散单位点PEG基团对活性造成的影响,
进一步提高PEG化干扰素的体外活性;2)更有效的封闭蛋白酶作用位点,更有效的提高干扰素的体内稳定性。 发明人选取上述筛选得到的H34及E107位点对干扰素进行双点的非天然氨基酸插入及PEG修饰。主要基于以下几点:1)此双点在上述实施实例中展现出较好的体外活性;2)此双点的非天然氨基酸插入效率较高;3)此双点对应着不同的受体结合区域。 发明人首先对双位点PEG修饰的可能性进行了鉴定,通过将此两个点突变有非天然氨基酸的干扰素分别于5K及20K的DIBO-PEG作用,经SDS-PAGE及考马斯亮蓝染色后,相应条带位置高于单位点插入非天然氨基酸干扰素与相应PEG反应产物,而与总PEG大小修饰产物位置相当(图4-A)。即双位点5K修饰反应物等于单位点10K修饰反应物;双位点20K修饰反应物高于单位点20K,却低于单位点40K,此与PEG分子类型有关。随后,发明人对双位点修饰PEG进行了圆二色谱的鉴定,确定了双位点PEG的修饰对蛋白质的二级结构没有造成明显的影响(图4-B)。 体外的胰酶稳定性试验可以简单有效的鉴别蛋白质对胰酶的抵抗能力,其效果可类比到体内。具体做法如下:样品同意稀释至0.4ug/ul,胰酶溶解至0.16ug/ul,将32ul样品与8ul胰酶混合,37℃混合作用,在不同时间点(1、2、3、4小时)加入10ul上样缓冲液,99℃煮沸记为终止。随后对样品进行SDS-PAGE电泳及考马斯亮蓝染色,并对结果进行灰度扫描分析,以确定各时间点样品的残留量。具体结果如图5所示。 由结果可知,H34&E107双点5K的修饰产物相比于单点10K修饰的干扰素而言,更有利于抵抗胰酶造成的蛋白酶解作用,其效果更优于单点5K的PEG修饰产物。双点5K的PEG修饰产物在胰酶作用4h后仍有部分残留,而其余样品在3小时以内基本降解完全。 随后发明人对双点修饰PEG的干扰素进行了体外活性的评价,SPR结果及体外的抗肿瘤、抗病毒活性数据总结如表4、表5:
表4.受体结合能力数据总结(SPR) 表5.双点PEG修饰体外抗肿瘤、抗病毒活性数据总结 *与WT数据归一化、以比较不同批实验数据(备注:体外活性每次试验的偏差会较大,若为不同批次测定的数据,应该与每一批次设定的标准组,即WT组均一化,各组数据才有比较性) 由表4可以看出,双位点修饰5K及20K的干扰素,其与受体结合能力低于或近似于单位点5K及20K的PEG化干扰素,而明显高于单位点10K及40K的PEG化干扰素。证明了双位点的较小PEG修饰有利于分散大分子量PEG对受体结合的影响。 表5表明,在体外活性上,双位点5K修饰的PEG化干扰素,其抗病毒活性低于单位点5K,而优于单位点10K;双位点20K修饰的PEG化干扰素,其活性近似于单位点40K;此结果一方面证明了双位点修饰存在着一定的优势、一方面也反应出在双位点修饰中,修饰位点及PEG分子量的选择仍存在很大灵活性。 实施例6:干扰素及其PEG化产物的体内药代动力学 为了评价PEG修饰对干扰素稳定性带来的影响,发明人对34位点不同PEG分子量修饰产物进行了药代动力学的体内试验。静脉注射曲线如图6-A所示、皮下注射曲线如图6-B所示;药代动力学参数如表6、表7所示: 表6.静脉注射药代动力学参数总结 表7.皮下注射药代动力学参数总结 由以上数据可以看出,对34位点的修饰而言,10K与20K的PEG修饰尽管在体外活性上有着相似的影响,但是H34-20K-IFN相比于H34-10K-IFN,其体内半衰期提高了将近一倍,清除率(Cl)及消除速率常数(Elimination rate)也有显著降低;而在H34及E107双位点20K-PEG修饰的干扰素虽然在体外活性上降低到单点40K修饰的活性,但是体内稳定性也近似或高于单点40K修饰的干扰素,提示多点的PEG修饰对提高体内稳定性是有显著作用的。 综合来看,数据显示,利用非天然氨基酸技术,在干扰素的34位点单点修饰PEG得到了最好的效果,H34-10K-IFN的体外抗病毒活性相当于未修饰的PEG的约40%,而对比上市药物PEG-INTRON约为25%(表2所示);在相同位点修饰20K-PEG的干扰素产品,即H34-20K-IFN,相比于10K的修饰,其保留了10K-PEG修饰约83%的体外抗病毒活性,据此我们认为在此位点20K-PEG的修饰相对10K而言,对活性影响不大(表3所示),而药代数据显示,相对于H34-10K-IFN,H34-20K-IFN的体内稳定性将近提高了两倍(表6、7中,T1/2及MRT所示);对于H34以及E107双位点PEG的修饰,我们实验的意图,在于通过多位点的小分子量PEG修饰,从而分散单位点大分子量PEG修饰对干扰素与其受体的相互作用影响,进而在不影响其体内稳定性的前提下(这里相对于单位点修饰较大PEG干扰素而言),进一步提高干扰素的保留活性。我们尝试的H34&E107-5K干扰素修饰产物,在体外的抗病毒活性上,其近似等于或高于H34-10K-IFN,而显著高于E107-10K-IFN(表5所示),而其体内的稳定性却近似等于单点修饰10K的干扰素(表6、7所示),此结果仅以两个位点及5K及20K的PEG做为尝试,并取得了预期的成果;而过程中我们也得到了有着不错性质的若干候补产物,例如在E107以及A74位点修饰有10K大小PEG的干扰素产品,都可作为进一步研究的候选产物。 虽然用上述实施方式描述了本发明,应当理解的是,在不背
离本发明的精神的前提下,本发明可进行进一步的修饰和变动,且这些修饰和变动均属于本发明的保护范围之内。例如,在双修饰干扰素的问题上,虽然本申请以H34及E107位点为例进行了说明,但是很明显,位点的选择不仅仅限于此两点,本发明所涉及的所有突变位点,及本发明未选择的,干扰素的其它位点都可适用。
Provided is a human interferon, wherein at least one amino acid residue is replaced with Lys-azido. Further provided is a pegylated human interferon, which improves in vivo stability of the human interferon. Additionally, provided is a method for preparing the interferon and a pegylated product thereof, which comprises using a plasmid which can express tRNAPyl/PylRS. 特定位点定点突变的人干扰素,其至少1个位点上的氨基酸被突变为非天然氨基酸,所述非天然氨基酸为: 所示的NAEK。 如权利要求1所述的定点突变人干扰素,其与突变前的干扰素的氨基酸序列的区别在于:氨基酸序列的第N位的氨基酸被突变为Lys-azido,突变氨基酸在干扰素中的连接方式,如下式所示: 其中,由R1到R2的方向为氨基酸序列的N末端到C末端方向,R1为干扰素的第1至第N-1位氨基酸残基; R2为干扰素的N+1位至C末端的氨基酸残基; R4为 如权利要求1或2所述的特定位点突变的人干扰素,其中未经突变的人干扰素序列如SEQ ID NO:1所示。 如权利要求3所述的特定位点突变的人干扰素,突变位点可为SEQ ID NO:1中任意位点上一个或多个的氨基酸。
如权利要求3所述的特定位点突变的人干扰素,所述突变位点选自:示于SEQ ID NO:1的序列的第P4位,H7位,S8位,K31位,R33位,H34位,E41位,E51位,A74位,E78位,G102位,T106位,E107位,L110位,M111位,E113位,L128位,K133位,P137位,E159位H34和E107位,或其他对活性影响较小的位点,以及多位点的组合。 如权利要求5所述的特定位点突变的人干扰素,所述突变位点选自:示于SEQ ID NO:1的序列的A74,H34,E107或其组合。 如权利要求5所述的特定位点突变的人干扰素,序列如SEQ ID NO:3、SEQ ID NO:4、SEQ ID NO:5或SEQ ID NO:6所示。 编码权利要求1-7中任一项的突变的干扰素的核酸分子。 如权利要求8所述的核酸分子,其中突变位点的氨基酸密码子被取代为琥珀密码子TAG。 核酸载体,其可操作地连接有权利要求8或9的核酸分子。 宿主细胞,其中含有权利要求10的核酸载体。 根据权利要求11的宿主细胞,其中还含有能够表达tRNAPyl/PylRS的质粒。 经PEG修饰的如权利要求1-7中任一项所述的特定位点突变的人干扰素,
其中,R1为突变前的干扰素的第1至第N-1位氨基酸残基, R2为突变的干扰素的第N+1位至C末端的氨基酸残基, R3为偶联的PEG分子。 如权利要求13所述的经PEG修饰的人干扰素,其特征在于当取代的氨基酸位点多于一个时,不同的取代位点偶联的PEG分子量可以是相同的,也可以是不同的。 如权利要求14所述的经PEG修饰的人干扰素,其中不同的取代位点偶联的PEG分子量是相同的。 经PEG修饰的如权利要求1-7中任一项所述的特定位点突变的人干扰素,其在非天然氨基酸位置定点引入PEG修饰,PEG的分子量范围为2kD-100kD。 如权利要求16所述的经PEG修饰的特定位点突变的人干扰素,所述PEG的分子量是5kD、10kD、20kD或者40kD。 如权利要求17所述的经PEG修饰的特定位点突变的人干扰素,具体是H34-5K-IFN、H34-10K-IFN、H34-20K-IFN、H34-40K-IFN、H34&E107-5K、H34&E107-20K、A74-5K-IFN、
A74-10K-IFN、A74-20K-IFN、A74-40K-IFN、E41-10K-IFN、E51-10K-IFN、E107-5K-IFN、E107-10K-IFN、E107-20K-IFN、A74-40K-IFN 如权利要求17或18所述的经PEG修饰的人干扰素,其特征在于当取代的氨基酸位点多于一个时,不同的取代位点偶联的PEG分子量可以是相同的,也可以是不同的。 如权利要求19所述的经PEG修饰的人干扰素,其中不同的取代位点偶联的PEG分子量是相同的。 药物组合物,其中含有有效量的权利要求1-7中任一项的干扰素或者权利要求13-20中任一项所述的经PEG修饰的干扰素,以及药学上可以接受的载体。 权利要求1-7中任一项的干扰素或者权利要求13-20中任一项所述的经PEG修饰的干扰素的制备方法,所述方法包括使用基因密码子扩展技术将非天然氨基酸定点引入干扰素蛋白中,借助非天然氨基酸上的特定基团和任选的修饰剂如聚乙二醇与干扰素定点连接;优选地,在干扰素中引入叠氮基团,再与含有环辛炔的PEG修饰剂进行无铜点击化学定点偶联PEG,用以制备PEG化干扰素。 权利要求1-7中任一项的干扰素或者权利要求13-20中任一项所述的经PEG修饰的干扰素在制备用于抗病毒,治疗多种恶性肿瘤,免疫调节的药物中的用途。
技术领域
背景技术
附图说明: