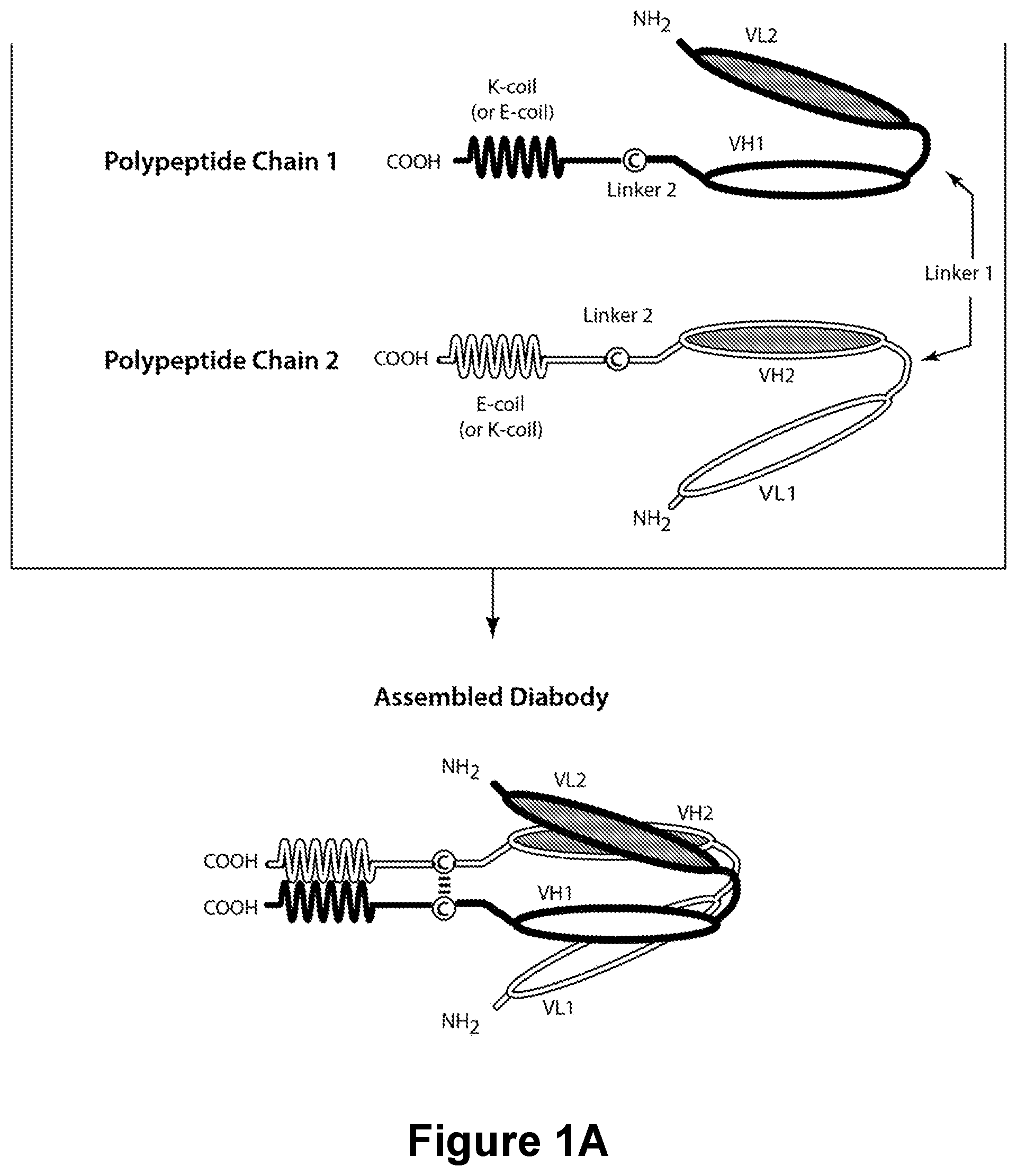
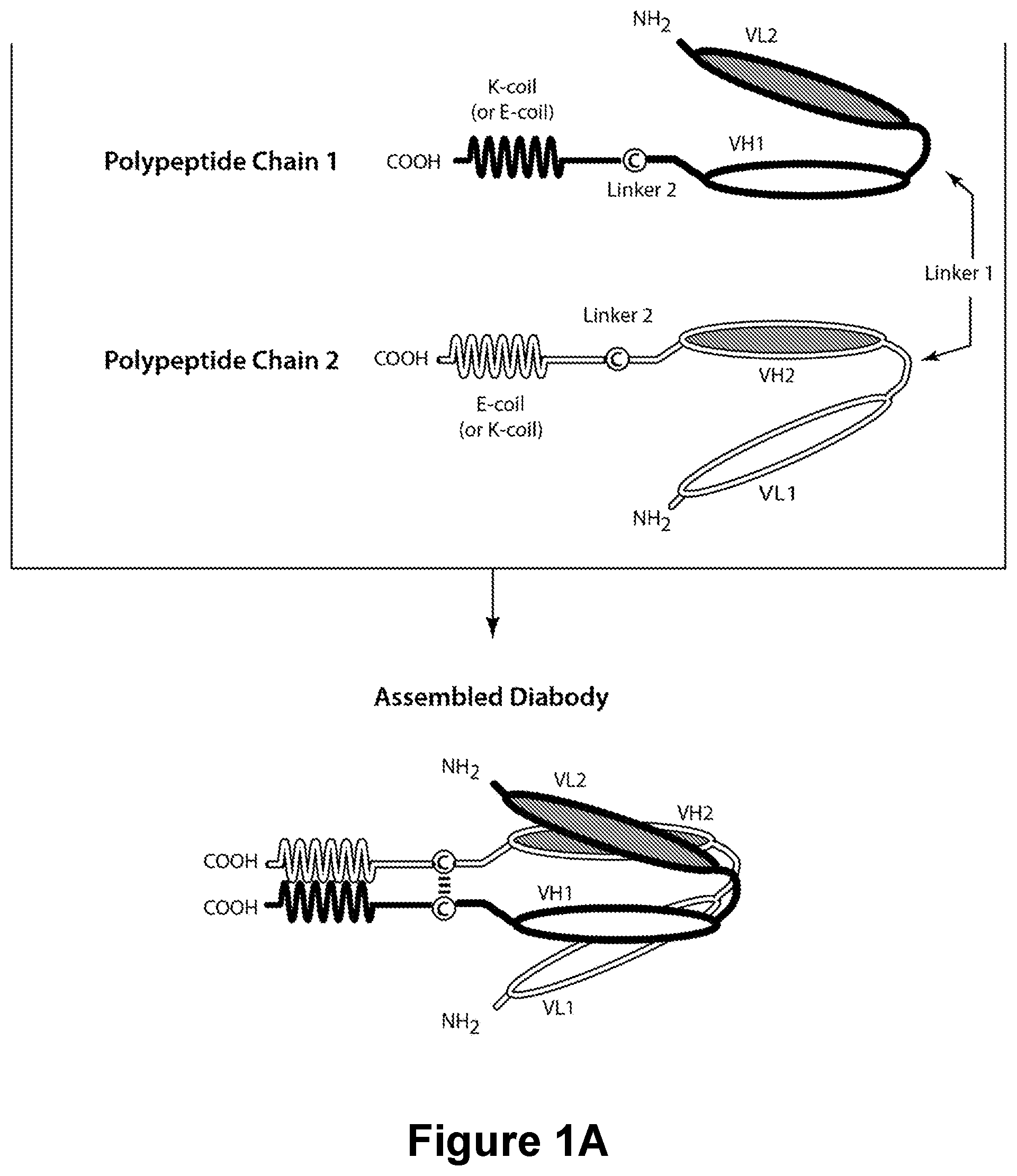
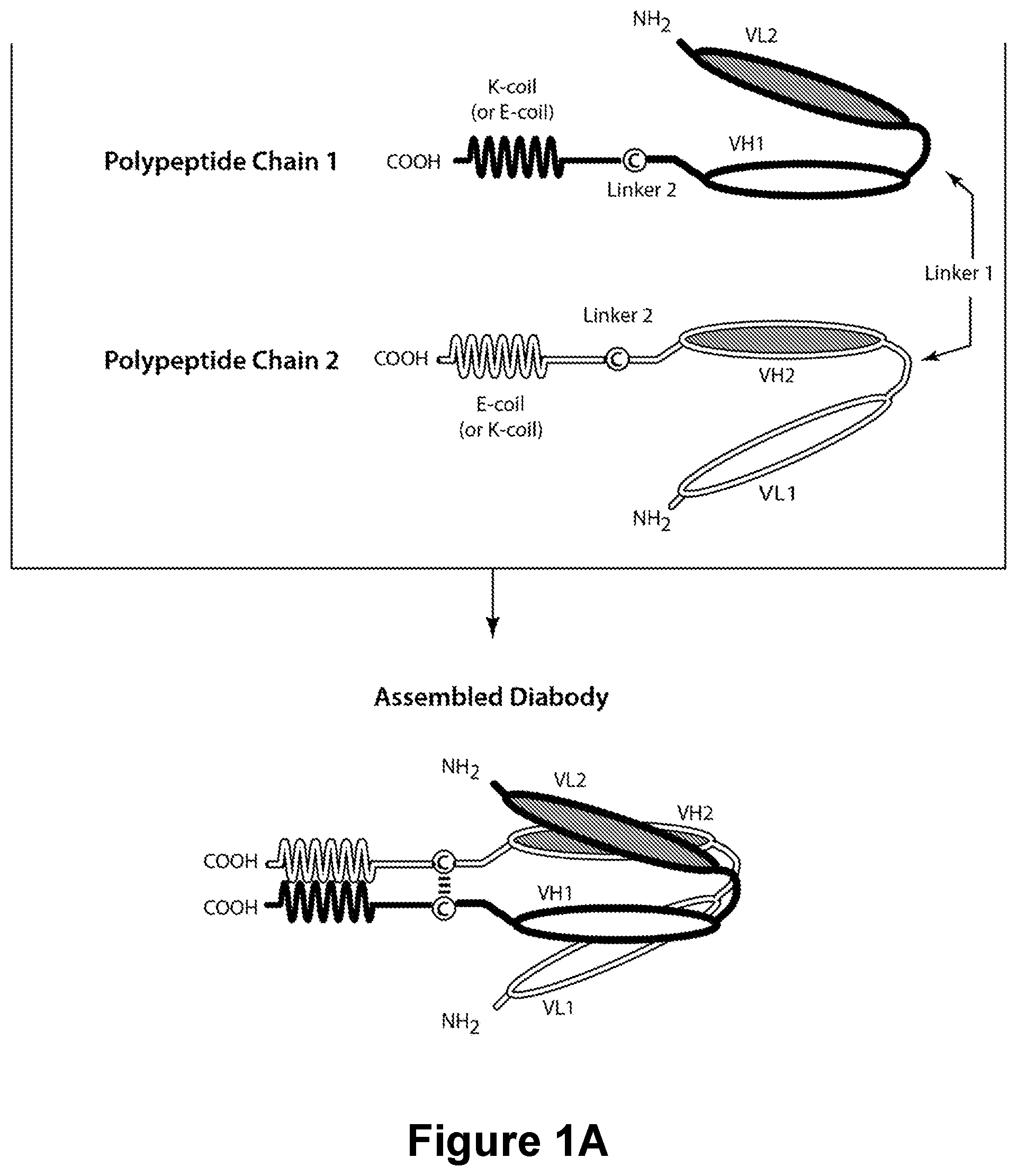
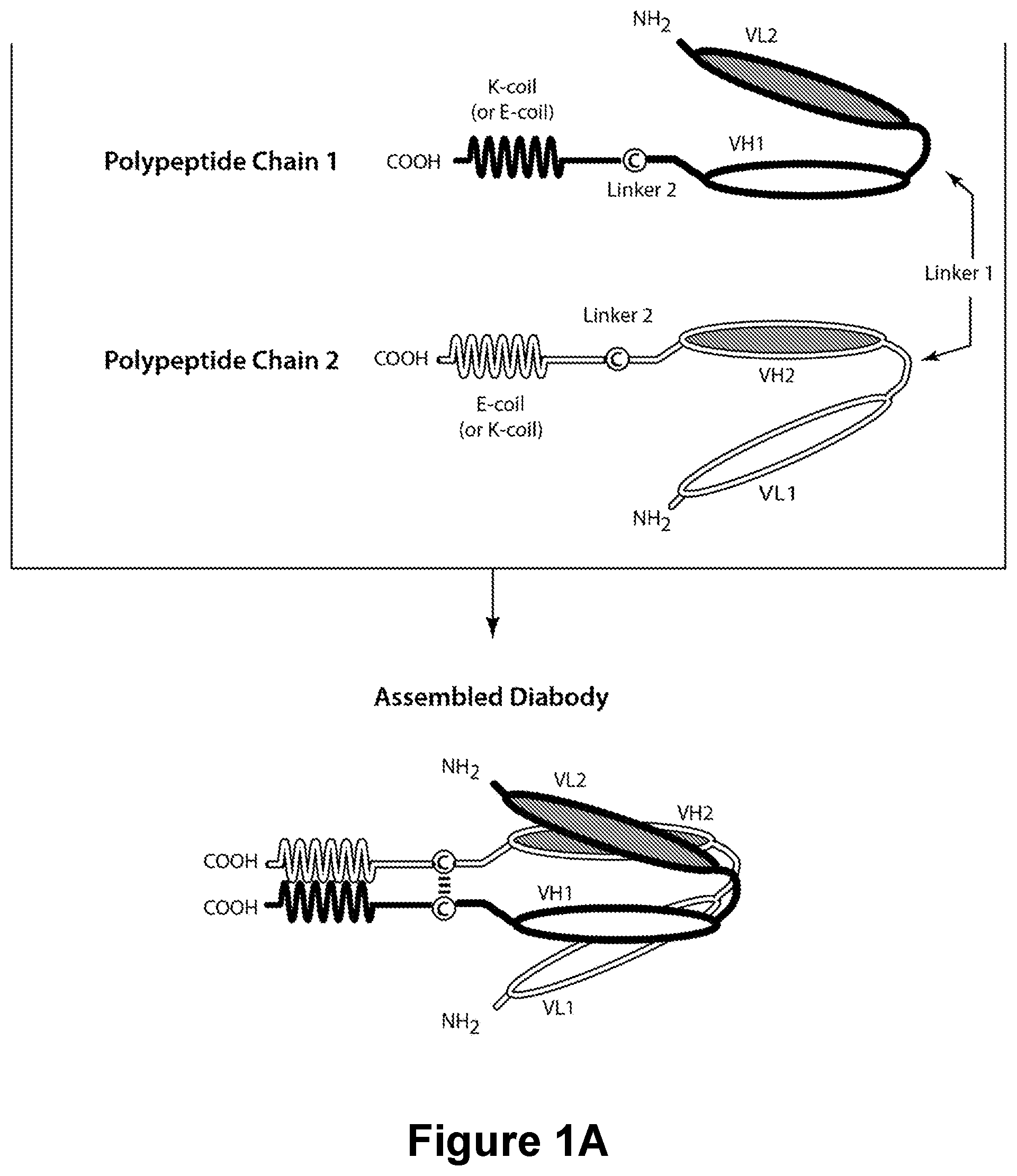
Bispecific CD16-Binding Molecules and Their Use in the Treatment of Disease
This application claims priority to U.S. Patent Appln. Ser. No. 62/597,800 (filed on Dec. 12, 2017; pending), which application is herein incorporated by reference in its entirety. This application includes one or more Sequence Listings pursuant to 37 C.F.R. 1.821 et seq., which are disclosed in computer-readable media (file name: 1301_0153PCT_Sequence_Listing_ST25.txt, created on Dec. 4, 2018, and having a size of 276,384 bytes), which file is herein incorporated by reference in its entirety. The present invention is directed to molecules (e.g., an antibody, a diabody, an scFv, an antibody, a TandAb, etc.) capable of binding an epitope of human CD16 (a “CD16 Binding Molecule”). The present invention is further directed to CD16 Binding Molecules that are capable of binding an epitope of human CD16 and one or more epitope(s) of a Disease Antigen (“DA”) (e.g., a “CD16 x DA Binding Molecule”). The present invention is particularly directed to such CD16 x DA Binding Molecules that are antibodies, or that comprise an Epitope Binding Domain thereof, or are diabodies (including DART® diabodies), bispecific antibodies, TandAbs, other multispecific binding molecules (e.g., trivalent TRIDENT™ molecules), etc. The invention particularly concerns CD16 x DA Binding Molecules that are capable of binding a Disease Antigen that is a Cancer Antigen or a Pathogen-Associated Antigen in addition to being able to bind CD16. The invention particularly concerns the use of such CD16 and CD16 x DA Binding Molecules in the treatment of cancer and pathogen-associated diseases. The present invention is also directed to pharmaceutical compositions that comprise such molecule(s). The mammalian immune system serves as a defense against a variety of conditions, including, e.g., injury, infection and neoplasia. The efficiency with which humans and other mammals develop an immunological response to pathogens, foreign substances and cancer antigens rests on two characteristics: the exquisite specificity of the immune response for antigen recognition, and the immunological memory that allows for faster and more vigorous responses upon re-activation with the same antigen (Portolés, P. et al. (2009) “ In healthy individuals, the immune system is in a quiescent state, inhibited by a repertoire of diverse inhibitory receptors and receptor ligands. Upon recognition of a cancer antigen, microbial pathogen, or an allergen, an array of activating receptors and receptor ligands are triggered to induce the activation of the immune system. Such activation leads to the activation of macrophages, Natural Killer (NK) cells and antigen-specific, cytotoxic, T-cells, and promotes the release of various cytokines, all of which act to counter the perceived threat to the health of the subject (Dong, C. et al. (2003) “ Thus, the disease state of cancer (and indeed the disease states of infectious diseases) may be considered to reflect a failure to adequately activate a subject's immune system. Such failure may reflect an inadequate presentation of activating immune signals, or it may reflect an inadequate ability to alleviate inhibitory immune signals in the subject. In some instances, researchers have determined that cancer cells can co-opt the immune system to evade being detected by the immune system (Topalian, S. L. et al. (2015) “Immune Checkpoint Blockade: Among the receptors involved in the activation of the immune system are the Fc Receptors:CD16, CD32 and CD64. These Fc receptors are found on the surfaces of multiple types of immune system cells (e.g., B lymphocytes, follicular dendritic cells, natural killer cells, macrophages, neutrophils, eosinophils, basophils and mast cells). Such receptors have an “extracellular” portion (which is thus capable of ligating to an Fc Domain), a “transmembrane” portion (which extends through the cellular membrane), and a “cytoplasmic” portion (positioned inside the cell). Multiple types of FcγRs have been identified: CD16A (FcγRIIIA), CD16B (FcγRIIIB), CD32A (FcγRIIA), CD32B (FcγRIIB), and CD64 (FcγRI). These receptors bind the Fc portion of IgG antibodies, thereby triggering the transduction of activating or inhibitory signals to the immune system. CD16 is a generic name for the activating Fc receptors, FcγRIIIA (CD16A) and FcγRIIIB (CD16B). These receptors bind the Fc portion of IgG antibodies, thereby triggering the release of cytokines. If such antibodies are bound to a Disease Antigen that is expressed on the surface of a cell (e.g., a cancer cell, pathogen-infected cell, etc.), then such release mediates the killing of the targeted cell. CD16A is expressed by Natural Killer (NK) cells and tissue macrophages that bind aggregated but not monomeric human IgG (Selvaraj, P. et al. (2004) “ The expression of CD16A by Natural Killer (NK) cells has particular relevance to the present invention, since such cells release cytokines when their CD16 molecules bind to the Fc Domain of an antibody. Thus, when a natural antibody binds to a Disease Antigen of a target cell, its Fc Domain can be recognized by a CD16 molecule of a Natural Killer cell, which then mediates the killing of the target cell. Since such killing is antibody-dependent, it is termed antibody-dependent cell-mediated cytotoxicity (ADCC). ADCC thus depends on a prior antibody response, and, as stated, requires the presence and participation of an FcγR-expressing effector cell (typically natural killer (NK) cells, but also macrophages, neutrophils and eosinophils). CD16A signals ADCC through interactions with the CD ξ protein of NK cells, and signals macrophage-mediated killing through interactions with macrophage FcγR chains. CD16A possesses two major polymorphic forms, F158 and V158, which differ by possessing a phenylalanine or a valine at residue 158 (shown as residue 160 of the extracellular domain of CD16A ( CD16B is expressed on neutrophils, and is anchored to glycophosphatidylinositol (“GPI-anchored”) (Meknache, N. et al. (2009) “ CD16B possesses two major polymorphic forms, NA1 and NA2, which exhibit different binding affinities for IgG1 and IgG3 subclasses (Bournazos, S. et al. (2010) “ CD32A (FcγRIIA) (Brandsma, A. M. (2015) “ The ability of the different FcγRs to mediate diametrically opposing functions reflects their structural differences, and in particular whether the FcγR possesses an immunoreceptor tyrosine-based activation motif (“ITAM”) or an immunoreceptor tyrosine-based inhibitory motif (“ITIM”). The recruitment of different cytoplasmic enzymes to these structures dictates the outcome of the FcγR-mediated cellular responses. ITAM-containing FcγRs include FcγRT, FcγRIIA, FcγRIIIA, and activate the immune system when bound to Fc Domains (e.g., aggregated Fc Domains present in an immune complex). FcγRIIB is the only currently known natural ITIM-containing FcγR; it acts to dampen or inhibit the immune system when bound to aggregated Fc Domains. Although natural IgG antibodies directed to an epitope of a particular Disease Antigen possess Fc Domains that can interact with CD16 molecules to activate a subject's immune response, in the case of many diseases and many subjects, such immune system activation is not sufficient to provide an effective therapy for the disease. Thus, despite prior advances in identifying the molecules involved in mammalian immune responses, a need remains for improved therapies for treating cancers and infectious diseases. The CD16-Binding Molecules of the present invention, and particularly, the CD16 x DA Binding Molecules of the present invention that comprise a CD16 Binding Domain and a Binding Domain specific for a Disease Antigen expressed on a target cell are capable of co-localizing CD16-expressing cells to the site(s) of cells expressing the Disease Antigen. Such co-localization enhances the ADCC-mediated killing of target cells by increasing the likelihood that an Fc portion of an antibody directed against an epitope of the Disease Antigen will bind to a CD16-expressing effector cell and, via such Fc-CD16 interaction, trigger immune system activation and the release of cytokines against the target cell. Thus, the present invention is directed to improving the activation of a subject's immune response to a Disease antigen and other goals. The present invention is directed to molecules (e.g., an antibody, a diabody, an scFv, an antibody, a TandAb, etc.) capable of binding an epitope of human CD16 (a “CD16 Binding Molecule”). The present invention is further directed to CD16 Binding Molecules that are capable of binding an epitope of human CD16 and one or more epitope(s) of a Disease Antigen (“DA”) (e.g., a “CD16 x DA Binding Molecule”). The present invention is particularly directed to such CD16 x DA Binding Molecules that are antibodies, or that comprise an Epitope Binding Domain thereof, or are diabodies (including DART® diabodies), bispecific antibodies, TandAbs, other multispecific binding molecules (e.g., trivalent TRIDENT™ molecules), etc. The invention particularly concerns CD16 x DA Binding Molecules that are capable of binding a Disease Antigen that is a Cancer Antigen or a Pathogen-Associated Antigen in addition to being able to bind CD16. The invention particularly concerns the use of such CD16 and CD16 x DA Binding Molecules in the treatment of cancer and pathogen-associated diseases. The present invention is also directed to pharmaceutical compositions that comprise such molecule(s). In detail, the invention provides a CD16 x Disease Antigen (CD16 x DA) Binding Molecule comprising a CD16 Binding Domain capable of binding an epitope of CD16 and also a Disease Antigen-Binding Domain capable of binding an epitope of a Disease Antigen, wherein the CD16 Binding Domain comprises one or more of:
The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecule, wherein the Molecule is a bispecific antibody, a bispecific diabody, a bispecific TandAb, a bispecific trivalent molecule, or a bispecific CAR. The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the Molecule is capable of binding more than one Disease Antigen and/or more than one epitope of CD16. The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the CD16 Binding Domain comprises:
The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the CD16 Binding Domain comprises:
The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the CD16 Binding Domain comprises:
The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the CD16 Binding Domain comprises:
The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the Disease Antigen is a Cancer Antigen and the disease is cancer. The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the cancer is selected from the group consisting of adrenal cancer, bladder cancer, breast cancer, colorectal cancer, gastric cancer, glioblastoma, kidney cancer, non-small-cell lung cancer, acute lymphocytic leukemia, acute myeloid leukemia, chronic lymphocytic leukemia, chronic myeloid leukemia, hairy cell leukemia, Burkett's lymphoma, diffuse large B cell lymphoma, follicular lymphoma, mantle cell lymphoma, marginal zone lymphoma, non-Hodgkin's lymphoma, small lymphocytic lymphoma, multiple myeloma, melanoma, ovarian cancer, pancreatic cancer, prostate cancer, skin cancer, renal cell carcinoma, testicular cancer, and uterine cancer. The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the Cancer Antigen is selected from the group consisting of the Cancer Antigens: 19.9, 4.2, A33, ADAM-9, AH6, ALCAM, B1, B7-H3, BAGE, beta-catenin, blood group ALeb/Ley, Burkitt's lymphoma antigen-38.13, C14, CA125, Carboxypeptidase M, CD5, CD19, CD20, CD22, CD23, CD25, CD27, CD28, CD33, CD36, CD40/CD154, CD45, CD56, CD46, CD52, CD56, CD79a/CD79b, CD103, CD123, CD317, CDK4, CEA, CEACAM5/CEACAM6, CO17-1A, CO-43, CO-514, CTA-1, CTLA-4, Cytokeratin 8, D1.1, Di56-22, DR5, E1series, EGFR, an Ephrin receptor, EphA2, Erb, GAGE, a GD2/GD3/GM2 ganglioside, GICA 19-9, gp100, Gp37, gp75, gpA33, HER2/neu, HMFG, human papillomavirus-E6/human papillomavirus-E7, HMW-MAA, I antigen, IL13Rα2, Integrin (36, JAM-3, KID3, KID31, KS 1/4 pan-carcinoma antigen, L6,L20, LEA, LUCA-2, M1:22:25:8, M18, M39, MAGE, MART, mesothelin, MUC-1, MUM-1, Myl, N-acetylglucosaminyltransferase, neoglycoprotein, NS-10, OFA-1, OFA-2, Oncostatin M, p15, p97, PEM, PEMA, PIPA, PSA, PSMA, prostatic acid phosphate, R24, ROR1, a sphingolipid, SSEA-1, SSEA-3, SSEA-4, sTn, the T cell receptor derived peptide, T5A7, TAG-72, TL5, TNF-receptor, TNF-γ receptor, TRA-1-85, a Transferrin Receptor, 5T4, TSTA, VEGF, a VEGF Receptor, VEP8, VEP9, VIM-D5, and Y hapten, Ley. The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the Disease Antigen is 5T4, B7-H3, CEACAM5/CEACAM6, CD19, CD123, EGRF, EphA2, HER2/neu, IL13Rα2 or VEGF. The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the Disease Antigen is a Pathogen-Associated Antigen. The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the Pathogen-Associated Antigen is selected from the group consisting of the Pathogen-Associated Antigens: Herpes Simplex Virus infected cell protein (ICP)47, Herpes Simplex Virus gD, Epstein-Barr Virus LMP-1, Epstein-Barr Virus LMP-2A, Epstein-Barr Virus LMP-2B, Human Immunodeficiency Virus gp160, Human Immunodeficiency Virus gp120, Human Immunodeficiency Virus gp41, etc.), Human Papillomavirus E6, Human Papillomavirus E7, human T-cell leukemia virus gp64, human T-cell leukemia virus gp46, and human T-cell leukemia virus gp21. The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the Disease Antigen is an HIV env antigen. The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the molecule is:
The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the molecule comprises an Fc Region. The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the Fc Region, is of the IgG1, IgG2, IgG3, or IgG4 isotype. The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules, wherein the Fc Region is a variant Fc Region that comprises:
The invention additionally concerns the embodiment of such CD16 x Disease Antigen Binding Molecules:
The invention additionally concerns a CD16 Binding Molecule, that comprises:
The invention additionally concerns the embodiment of such a CD16 Binding Molecule wherein the molecule comprises:
The invention additionally concerns a CD16 Binding Molecule that comprises:
The invention additionally concerns the embodiment of such CD16 Binding Molecule wherein the molecule comprises:
The invention additionally concerns the embodiment of such CD16 Binding Molecules wherein the molecule is selected from the group consisting of: an antibody, a multispecific antibody, a Fab′ fragment, a F(ab′)2 fragment, a (Fv) fragment, a single-chain (scFv), a single-chain antibody, a disulfide-linked bispecific Fv (sdFv), a diabody, a trivalent binding molecule, and a CAR-T molecule. The invention additionally concerns a pharmaceutical composition that comprises any of the above-described CD16 x Disease Antigen Binding Molecules, or CD16 Binding Molecules, and a pharmaceutically acceptable carrier. The invention additionally concerns the use of the above-described pharmaceutical composition in the treatment of a disease characterized by the expression of the Disease Antigen. The invention additionally concerns a method for the treatment of a disease characterized by the expression of the Disease Antigen, comprising administering to a subject in need thereof a therapeutically effective amount of the above-described pharmaceutical composition. The invention additionally concerns the embodiment of such use or method wherein the CD16 x Disease Antigen Binding Molecule is capable of binding more than one Disease Antigen and/or more than one epitope of CD16. The invention additionally concerns such use or method wherein the CD16 x Disease Antigen Binding Molecule, wherein the Disease Antigen is a Cancer Antigen, and the disease is cancer. The invention additionally concerns such use or method wherein the cancer is selected from the group consisting of adrenal cancer, bladder cancer, breast cancer, colorectal cancer, gastric cancer, glioblastoma, kidney cancer, non-small-cell lung cancer, acute lymphocytic leukemia, acute myeloid leukemia, chronic lymphocytic leukemia, chronic myeloid leukemia, hairy cell leukemia, Burkett's lymphoma, diffuse large B cell lymphoma, follicular lymphoma, mantle cell lymphoma, marginal zone lymphoma, non-Hodgkin's lymphoma, small lymphocytic lymphoma, multiple myeloma, melanoma, ovarian cancer, pancreatic cancer, prostate cancer, skin cancer, renal cell carcinoma, testicular cancer, and uterine cancer. The invention additionally concerns such use or method wherein the Cancer Antigen is selected from the group consisting of the Cancer Antigens: 19.9, 4.2, A33, ADAM-9, AH6, ALCAM, B1, B7-H3, BAGE, beta-catenin, blood group ALeb/Ley, Burkitt's lymphoma antigen-38.13, C14, CA125, Carboxypeptidase M, CD5, CD19, CD20, CD22, CD23, CD25, CD27, CD28, CD33, CD36, CD40/CD154, CD45, CD56, CD46, CD52, CD56, CD79a/CD79b, CD103, CD123, CD317, CDK4, CEA, CEACAM5/CEACAM6, CO17-1A, CO-43, CO-514, CTA-1, CTLA-4, Cytokeratin 8, D1.1, Di56-22, DR5, E1series, EGFR, an Ephrin receptor, EphA2, Erb, GAGE, a GD2/GD3/GM2 ganglioside, GICA 19-9, gp100, Gp37, gp75, gpA33, HER2/neu, HMFG, human papillomavirus-E6/human papillomavirus-E7, HMW-MAA, I antigen, IL13Rα2, Integrin (36, JAM-3, KID3, KID31, KS 1/4 pan-carcinoma antigen, L6,L20, LEA, LUCA-2, M1:22:25:8, M18, M39, MAGE, MART, mesothelin, MUC-1, MUM-1, Myl, N-acetylglucosaminyltransferase, neoglycoprotein, NS-10, OFA-1, OFA-2, Oncostatin M, p15, p97, PEM, PEMA, PIPA, PSA, PSMA, prostatic acid phosphate, R24, ROR1, a sphingolipid, SSEA-1, SSEA-3, SSEA-4, sTn, the T cell receptor derived peptide, T5A7, TAG-72, TL5, TNF-receptor, TNF-γ receptor, TRA-1-85, a Transferrin Receptor, 5T4, TSTA, VEGF, a VEGF Receptor, VEP8, VEP9, VIM-D5, and Y hapten, Ley. The invention additionally concerns such use or method wherein the Disease Antigen is 5T4, B7-H3, CEACAM5/CEACAM6, CD19, CD123, EGRF, EphA2, HER2/neu, IL13Rα2 or VEGF. The invention additionally concerns such use or method wherein the CD16 x Disease Antigen Binding Molecule, wherein the Disease Antigen is a Pathogen-Associated Antigen. The invention additionally concerns such use or method wherein the Pathogen-Associated Antigen is selected from the group consisting of the Pathogen-Associated Antigens: Herpes Simplex Virus infected cell protein (ICP)47, Herpes Simplex Virus gD, Epstein-Barr Virus LMP-1, Epstein-Barr Virus LMP-2A, Epstein-Barr Virus LMP-2B, Human Immunodeficiency Virus gp160, Human Immunodeficiency Virus gp120, Human Immunodeficiency Virus gp41, etc.), Human Papillomavirus E6, Human Papillomavirus E7, human T-cell leukemia virus gp64, human T-cell leukemia virus gp46, and human T-cell leukemia virus gp21. The invention additionally concerns such use or method wherein the Disease Antigen is an HIV env antigen. The present invention is directed to molecules (e.g., an antibody, a diabody, an scFv, an antibody, a TandAb, etc.) capable of binding an epitope of human CD16 (a “CD16 Binding Molecule”). The present invention is further directed to CD16 Binding Molecules that are capable of binding an epitope of human CD16 and one or more epitope(s) of a Disease Antigen (“DA”) (e.g., a “CD16 x DA Binding Molecule”). The present invention is particularly directed to such CD16 x DA Binding Molecules that are antibodies, or that comprise an Epitope Binding Domain thereof, or are diabodies (including DART® diabodies), bispecific antibodies, TandAbs, other multispecific binding molecules (e.g., trivalent TRIDENT™ molecules), etc. The invention particularly concerns CD16 x DA Binding Molecules that are capable of binding a Disease Antigen that is a Cancer Antigen or a Pathogen-Associated Antigen in addition to being able to bind CD16. The invention particularly concerns the use of such CD16 and CD16 x DA Binding Molecules in the treatment of cancer and pathogen-associated diseases. The present invention is also directed to pharmaceutical compositions that comprise such molecule(s). The CD16 x DA Binding Molecules of the present invention may be antibodies, or be derivable from antibodies (e.g., by fragmentation, cleavage, etc. of antibody polypeptides, or from use of the amino acid sequences of antibody molecules or of polynucleotides (or their sequences) that encode such polynucleotides, etc.). Antibodies are immunoglobulin molecules capable of specific binding to a particular domain or moiety or conformation (an “epitope”) of a molecule, such as a carbohydrate, polynucleotide, lipid, polypeptide, etc. An epitope-containing molecule may have immunogenic activity, such that it elicits an antibody production response in an animal; such molecules are termed “antigens.” As used herein, the terms “antibody” and “antibodies” refer to monoclonal antibodies, multispecific antibodies, human antibodies, humanized antibodies, synthetic antibodies, chimeric antibodies, polyclonal antibodies, camelized antibodies, single-chain Fvs (scFv), single-chain antibodies, Fab fragments, F(ab′) fragments, disulfide-linked bispecific Fvs (sdFv), intrabodies, and Epitope Binding Domains of any of the above. Such Immunoglobulin molecules can be of any type (e.g., IgG, IgE, IgM, IgD, IgA and IgY), class (e.g., IgG1, IgG2, IgG3, IgG4, IgA1and IgA2) or subclass. The term “monoclonal antibody” refers to a homogeneous antibody population wherein the monoclonal antibody is comprised of amino acids (naturally occurring or non-naturally occurring) that are involved in the selective binding of an antigen. Monoclonal antibodies are highly specific, being directed against a single epitope (or antigenic site). The term “monoclonal antibody” encompasses not only intact monoclonal antibodies and full-length monoclonal antibodies, but also fragments thereof (such as Fab, Fab′, F(ab′)2, (Fv), single-chain (scFv), mutants thereof, fusion proteins comprising an antibody portion, humanized monoclonal antibodies, chimeric monoclonal antibodies, and any other modified configuration of the immunoglobulin molecule that comprises an antigen recognition site of the required specificity and the ability to bind to an antigen. It is not intended to be limited as regards to the source of the antibody or the manner in which it is made (e.g., by hybridoma, phage selection, recombinant expression, transgenic animals, etc.). The term includes whole immunoglobulins as well as the fragments etc. described above under the definition of “antibody.” Methods of making monoclonal antibodies are known in the art. One method which may be employed is the method of Kohler, G. et al. (1975) “ Antibodies and the Binding Molecules of the present invention bind epitopes via their Binding Domains in an “immunospecific” manner. As used herein, a molecule is said to bind an epitope of another molecule in an immunospecific manner (or “immunospecifically”) if it binds or associates more frequently, more rapidly, with greater duration and/or with greater affinity with that epitope relative to alternative epitopes. For example, an antibody that immunospecifically binds to a viral epitope is an antibody that binds this viral epitope with greater affinity, avidity, more readily, and/or with greater duration than it immunospecifically binds to other viral epitopes or non-viral epitopes. It is also understood by reading this definition that, for example, an antibody (or moiety or epitope) that immunospecifically binds to a first target may or may not specifically or preferentially bind a second target. As such, “immunospecific binding” does not necessarily require (although it can include) exclusive binding. Generally, but not necessarily, reference to binding means “immunospecific” binding. Natural antibodies are capable of binding to only one epitope species (i.e., they are “monospecific”), although they can immunospecifically bind multiple copies of that species (i.e., exhibiting “bivalency” or “multivalency”). Two molecules are said to be capable of binding one another in a “physiospecific” manner, if such binding exhibits the specificity with which receptors bind their respective ligands. The last few decades have seen a revival of interest in the therapeutic potential of antibodies, and antibodies have become one of the leading classes of biotechnology-derived drugs (Chan, C. E. et al. (2009) “ The basic structural unit of naturally occurring immunoglobulins (e.g., IgG) is a tetramer composed of two shorter “Light Chains” complexed with two longer “Heavy Chains” and is usually expressed as a glycoprotein of about 150,000 Da. Each chain is composed of an amino-terminal (“N-terminal”) portion that comprises a “Variable Domain” and a carboxy-terminal (“C-terminal”) portion that comprises at least one “Constant Domain.” An IgG Light Chain is composed of a single “Light Chain Variable Domain” (“VL”) and a single “Light Chain Constant Domain” (“CL”). Thus, the structure of the light chains of an IgG molecule is n-VL-CL-c (where n and c represent, respectively, the N-terminus and the C-terminus of the polypeptide). An IgG Heavy Chain is composed of a single “Heavy Chain Variable Domain” (“VH”), three “Heavy Chain Constant Domains” (“CH1,” “CH2” and “CH3”), and a “Hinge” Region (“H”), located between the CH1 and CH2 Domains. Thus, the structure of an IgG heavy chain is n-VH—CH1-H—CH2-CH3-c (where n and c represent, respectively, the N-terminus and the C-terminus of the polypeptide). The ability of an intact, unmodified antibody (e.g., an IgG antibody) to bind an epitope of an antigen depends upon the presence and sequences of the Variable Domains. Unless specifically noted, the order of domains of the protein molecules described herein is in the “N-terminal to C-terminal” direction. A preferred CL Domain is a human IgG CL Kappa Domain. The amino acid sequence of an exemplary human CL Kappa Domain is (SEQ ID NO:1): Alternatively, an exemplary CL Domain is a human IgG CL Lambda Domain. The amino acid sequence of an exemplary human CL Lambda Domain is (SEQ ID NO:2): The CH1 Domains of the two Heavy Chains of an antibody complex with the antibody's Light Chain's “CL” constant region, and are attached to the Heavy Chains CH2 Domains via an intervening Hinge Domain. An exemplary CH1 Domain is a human IgG1 CH1 Domain. The amino acid sequence of an exemplary human IgG1 CH1 Domain is (SEQ ID NO:3): An exemplary CH1 Domain is a human IgG2 CH1 Domain. The amino acid sequence of an exemplary human IgG2 CH1 Domain is (SEQ ID NO:4): An exemplary CH1 Domain is a human IgG3 CH1 Domain. The amino acid sequence of an exemplary human IgG3 CH1 Domain is (SEQ ID NO:5): An exemplary CH1 Domain is a human IgG4 CH1 Domain. The amino acid sequence of an exemplary human IgG4 CH1 Domain is (SEQ ID NO:6): One exemplary Hinge Domain is a human IgG1 Hinge Domain. The amino acid sequence of an exemplary human IgG1 Hinge Domain is (SEQ ID NO:7): Another exemplary Hinge Domain is a human IgG2 Hinge Domain. The amino acid sequence of an exemplary human IgG2 Hinge Domain is (SEQ ID NO:8): Another exemplary Hinge Domain is a human IgG3 Hinge Domain. The amino acid sequence of an exemplary human IgG2 Hinge Domain is (SEQ ID NO:9): Another exemplary Hinge Domain is a human IgG4 Hinge Domain. The amino acid sequence of an exemplary human IgG4 Hinge Domain is (SEQ ID NO:10): The CH2 and CH3 Domains of the two heavy chains interact to form the “Fc Domain” of IgG antibodies that is recognized by cellular Fc Receptors, including but not limited to Fc gamma Receptors (FcγRs). As used herein, the term “Fc Region” is used to define a C-terminal region of an IgG heavy chain. A portion of an Fc Region (including a portion that encompasses an entire Fc Region) is referred to herein as an “Fc Domain.” An Fc Region is said to be of a particular IgG isotype, class or subclass if its amino acid sequence is most homologous to that isotype relative to other IgG isotypes. In addition to their known uses in diagnostics, antibodies have been shown to be useful as therapeutic agents. The amino acid sequence of the CH2-CH3 Domain of an exemplary human IgG1 is (SEQ ID NO:12): The amino acid sequence of the CH2-CH3 Domain of an exemplary human IgG2 is (SEQ ID NO:13): The amino acid sequence of the CH2-CH3 Domain of an exemplary human IgG3 is (SEQ ID NO:14): The amino acid sequence of the CH2-CH3 Domain of an exemplary human IgG4 is (SEQ ID NO:15): Throughout the present specification, the numbering of the residues in the constant region of an IgG heavy chain is that of the EU index as in Kabat et al., S Polymorphisms have been observed at a number of different positions within antibody constant regions (e.g., Fc positions, including but not limited to positions 270, 272, 312, 315, 356, and 358 as numbered by the EU index as set forth in Kabat), and thus slight differences between the presented sequence and sequences in the prior art can exist. Polymorphic forms of human immunoglobulins have been well-characterized. At present, 18 Gm allotypes are known: G1m (1, 2, 3, 17) or G1m (a, x, f, z), G2m (23) or G2m (n), G3m (5, 6, 10, 11, 13, 14, 15, 16, 21, 24, 26, 27, 28) or G3m (b1, c3, b3, b0, b3, b4, s, t, g1, c5, u, v, g5) (Lefranc et al., “ The Variable Domains of an IgG molecule consist of three “complementarity determining regions” (“CDRs”), which contain the amino acid residues of the antibody that will be in contact with the epitope, as well as intervening non-CDR segments, referred to as “framework regions” (“FRs”), which, in general maintain the structure and determine the positioning of the CDR loops so as to permit such contacting (although certain framework residues may also contact the epitope). Thus, the VL and VH Domains have the structure n-FR1-CDR1-FR2-CDR2-FR3-CDR3-FR4-c. The amino acid sequences of the CDRs determine whether an antibody will be able to bind to a particular epitope. Interaction of an antibody light chain with an antibody heavy chain and, in particular, interaction of their VL and VH Domains, forms an epitope-binding site of the antibody. Polypeptides that are (or may serve as) the first, second and third CDR of the Light Chain of an antibody are herein respectively designated as: CDRL1 Domain, CDRL2 Domain, and CDRL3 Domain. Similarly, polypeptides that are (or may serve as) the first, second and third CDR of the Heavy Chain of an antibody are herein respectively designated as: CDRH1 Domain, CDRH2 Domain, and CDRH3 Domain. Thus, the terms CDRL1 Domain, CDRL2 Domain, CDRL3 Domain, CDRH1 Domain, CDRH2 Domain, and CDRH3 Domain are directed to polypeptides that when incorporated into a protein cause that protein to be able to bind to a specific epitope regardless of whether such protein is an antibody having light and heavy chains or is a diabody or a single-chain binding molecule (e.g., an scFv, a BiTe, etc.), or is another type of protein. The term “Epitope Binding Domain” denotes a fragment or portion of a binding molecule (or a polypeptide having the amino acid sequence of such a fragment or portion) that contributes to the ability of the binding molecule to immunospecifically bind to an epitope. An Epitope Binding Domain may contain a VL or VH Domain of an antibody, or any 1, 2, 3, 4, or 5 of the CDR Domains of an antibody, or may contain all 6 of the CDR Domains of an antibody and, although capable of immunospecifically binding such epitope, may exhibit an immunospecificity, affinity or selectivity towards such epitope that differs from that of such antibody. An Epitope Binding Domain may contain only part of a CDR, namely the subset of CDR residues required for binding, termed the SDRs (Kim, J. H. et al. (2012) “ The invention also particularly encompasses Binding Molecules that comprise a VL or VH Domain of an antibody, and preferably both a VL and a VH Domain of an antibody. Preferably, such antibody is a humanized antibody. Monoclonal antibodies are typically prepared in non-human species, such as mouse or rabbit. The Variable and/or Constant Domains of such antibodies may be recognized as immunogens, thus provoking an immune response against them. Such molecules may however be “humanized” by introducing one or more amino acid substitutions in order to render such antibodies more like antibodies produced by humans. thereby reducing or eliminating their immunogenicity. The term “humanized” antibody refers to a chimeric molecule, generally prepared using recombinant techniques, having an epitope-binding site of an immunoglobulin from a non-human species and a remaining immunoglobulin structure of the molecule that is based upon the structure and/or sequence of a human immunoglobulin. The polynucleotide sequence of the variable domains of such antibodies may be used for genetic manipulation to generate such derivatives and to improve the affinity, or other characteristics of such antibodies. Application of this approach to various antibodies has been reported by LoBuglio, A. F. et al. (1989) “ The general principle in humanizing an antibody involves retaining the basic sequence of the Epitope Binding Domain of the antibody, while swapping the non-human remainder of the antibody with human antibody sequences. There are four general steps to humanize a monoclonal antibody. These are: (1) determining the nucleotide and predicted amino acid sequence of the starting antibody light and heavy variable domains (2) designing the humanized antibody or caninized antibody, i.e., deciding which antibody framework region to use during the humanizing or canonizing process (3) the actual humanizing or caninizing methodologies/techniques and (4) the transfection and expression of the humanized antibody. See, for example, U.S. Pat. Nos. 4,816,567; 5,807,715; 5,866,692; and 6,331,415. A number of humanized antibody molecules comprising an Epitope Binding Domain derived from a non-human immunoglobulin have been described, including chimeric antibodies having rodent or modified rodent Variable Domain and their associated complementarity determining regions (CDRs) fused to human constant domains (see, for example, Winter et al. (1991) “ Notwithstanding such successes, the production of stable, functional heterodimeric, non-monospecific diabodies optimized for therapeutic use can be further improved by the careful consideration and placement of the domains employed in the polypeptide chains. The present invention is thus directed to the provision of specific polypeptides that are particularly designed to form, via covalent bonding, stable and therapeutically useful heterodimeric diabodies and heterodimeric Fc diabodies that are capable of simultaneously binding CD16 and a Disease Antigen. As indicated above, natural antibodies are capable of binding to only one epitope species, although they can bind multiple copies of that species. The ability of an antibody to bind an epitope of an antigen depends upon the presence and amino acid sequence of the antibody's VL and VH Domains. Interaction of an antibody's Light Chain and Heavy Chain and, in particular, interaction of its VL and VH Domains forms one of the two Epitope Binding Domains of a natural antibody, such as an IgG. Natural antibodies are capable of binding only one epitope species (i.e., they are mono-specific), although they can bind multiple copies of that species (i.e., exhibiting bi-valency or multi-valency). The functionality of antibodies can be enhanced by generating multispecific antibody-based molecules that can simultaneously bind two separate and distinct antigens (or different epitopes of the same antigen) and/or by generating antibody-based molecule having higher valency (i.e., more than two Binding Domains) for the same epitope and/or antigen. In order to provide molecules having greater capability than natural antibodies, a wide variety of recombinant bispecific antibody formats have been developed (see, e.g., PCT Publication Nos. WO 2008/003116, WO 2009/132876, WO 2008/003103, WO 2007/146968, WO 2009/018386, WO 2012/009544, WO 2013/070565). Most of such approaches use linker peptides to fuse a further binding domain (e.g. an scFv, VL, VH, etc.) to, or within the antibody core (IgA, IgD, IgE, IgG or IgM), or to fuse multiple antibody binding portions to one another (e.g. two Fab fragments or scFv). Alternative formats use linker peptides to fuse a binding protein (e.g., an scFv, VL, VH, etc.) to a dimerization domain such as the CH2-CH3 Domain or alternative polypeptides (WO 2005/070966, WO 2006/107786A WO 2006/107617A, WO 2007/046893). Typically, such approaches involve compromises and trade-offs. For example, PCT Publication Nos. WO 2013/174873, WO 2011/133886 and WO 2010/136172 disclose that the use of linkers may cause problems in therapeutic settings, and teaches a trispecific antibody in which the CL and CH1 Domains are switched from their respective natural positions and the VL and VH Domains have been diversified (WO 2008/027236; WO 2010/108127) to allow them to bind to more than one antigen. Thus, the molecules disclosed in these documents trade binding specificity for the ability to bind additional antigen species. PCT Publication Nos. WO 2013/163427 and WO 2013/119903 disclose modifying the CH2 Domain to contain a fusion protein adduct comprising a binding domain. The document notes that the CH2 Domain likely plays only a minimal role in mediating effector function. PCT Publication Nos. WO 2010/028797, WO2010028796 and WO 2010/028795 disclose recombinant antibodies whose Fc Regions have been replaced with additional VL and VH Domains, so as to form trivalent binding molecules. PCT Publication Nos. WO 2003/025018 and WO2003012069 disclose recombinant diabodies whose individual chains contain scFv domains. PCT Publication Nos. WO 2013/006544 discloses multi-valent Fab molecules that are synthesized as a single polypeptide chain and then subjected to proteolysis to yield heterodimeric structures. Thus, the molecules disclosed in these documents trade all or some of the capability of mediating effector function for the ability to bind additional antigen species. PCT Publication Nos. WO 2014/022540, WO 2013/003652, WO 2012/162583, WO 2012/156430, WO 2011/086091, WO 2008/024188, WO 2007/024715, WO 2007/075270, WO 1998/002463, WO 1992/022583 and WO 1991/003493 disclose adding additional Binding Domains or functional groups to an antibody or an antibody portion (e.g., adding a diabody to the antibody's light chain, or adding additional VL and VH Domains to the antibody's light and heavy chains, or adding a heterologous fusion protein or chaining multiple Fab Domains to one another). Thus, the molecules disclosed in these documents trade native antibody structure for the ability to bind additional antigen species. The binding molecules of the present invention may be Chimeric Antigen Receptors (“CARs”) that comprise a single chain variable fragment (scFv) capable of binding CD16 and a Disease Antigen. As indicated above, scFvs are made by linking Light and Heavy Chain Variable Domains together via a short linking peptide. First-generation CARs typically had the intracellular domain from the CD3 chain, which is the primary transmitter of signals from endogenous TCRs. Second-generation CARs possessed additional intracellular signaling domains from various costimulatory protein receptors (e.g., CD28, 41BB, ICOS, etc.) to the cytoplasmic tail of the CAR in order to provide additional signals to the T-cell. Third-generation CARs combine multiple signaling domains, such as CD3z-CD28-41BB or CD3z-CD28-OX40, in order to further augment potency (Tettamanti, S. et al. (2013) “ The intracellular domain of the CARs of the present invention is preferably selected from the intracellular domain of any of: 41BB-CD3ζ, b2c-CD3ζ, CD28, CD28-4-1BB-CD3ζ, CD28-CD3ζ, CD28-FcεRIγ, CD28mut-CD3ζ, CD28-OX40-CD3ζ, CD28-OX40-CD3ζ, CD3, CD4-CD3ζ, CD4-FcεRIγ, CD8-CD3ζ, FcεRIγ, FcεRIγCAIX, Heregulin-CD3ζ, IL-13-CD3ζ, or Ly49H-CD3ζ (Tettamanti, S. et al. (2013) “ The art has additionally noted the capability of producing diabodies that differ from natural antibodies in being capable of binding two or more different epitope species (i.e., exhibiting bispecificity or multispecificity in addition to bi-valency or multi-valency) (see, e.g., Holliger et al. (1993) “′ The design of a diabody is based on the structure of the single-chain Variable Domain fragment (scFv), in which Light and Heavy Chain Variable Domains are linked to one another using a short linking peptide. Bird et al. (1988) (“ The provision of non-monospecific “diabodies” provides a significant advantage over antibodies: the capacity to co-ligate and co-localize cells that express different epitopes. Bispecific diabodies thus have wide-ranging applications including therapy and immunodiagnosis. Bispecificity allows for great flexibility in the design and engineering of the diabody in various applications, providing enhanced avidity to multimeric antigens, the cross-linking of differing antigens, and directed targeting to specific cell types relying on the presence of both target antigens. Due to their bivalency, low dissociation rates and rapid clearance from the circulation (for diabodies of small size, at or below ˜50 kDa), diabody molecules known in the art have also shown particular use in the field of tumor imaging (Fitzgerald et al. (1997) “ The ability to produce bispecific diabodies has led to their use (in “trans”) to co-ligate two cells together, for example, by co-ligating receptors that are present on the surface of different cells (e.g., cross-linking cytotoxic T-cells to target cells, such as cancer cells or pathogen-infected cells, that express a Disease Antigen) (Staerz et al. (1985) “ In many studies, diabody binding to effector cell determinants, e.g., Fcγ receptors (FcγR), was also found to activate the effector cell (Holliger et al. (1996) “ However, the advantages of the above-described bispecific diabodies come at a salient cost. The formation of such non-mono-specific diabodies requires the successful assembly of two or more distinct and different polypeptides (i.e., such formation requires that the diabodies be formed through the heterodimerization of different polypeptide chain species). This fact is in contrast to mono-specific diabodies, which are formed through the homodimerization of identical polypeptide chains. Because at least two dissimilar polypeptides (i. e., two polypeptide species) must be provided in order to form a non-mono-specific diabody, and because homodimerization of such polypeptides leads to inactive molecules (Takemura, S. et al. (2000) “ However, the art has recognized that bispecific diabodies composed of non-covalently associated polypeptides are unstable and readily dissociate into non-functional single polypeptide chain monomers (see, e.g., Lu, D. et al. (2005) “ In the face of this challenge, the art has succeeded in developing stable, covalently bonded heterodimeric non-mono-specific diabodies, termed DART® diabodies, see, e.g., Sloan, D. D. et al. (2015) “ The simplest DART® diabody comprises two polypeptide chains each comprising three Domains ( Alternative constructs are known in the art for applications where a bispecific or tetravalent molecule is desirable but an Fc is not required including, but not limited to, Bispecific T cell Engager molecules, also referred to as “BITE® antibodies” (see, e.g., PCT Publication Nos: WO 1993/11161; and WO 2004/106381) and tetravalent tandem antibodies, also referred to as “TandAbs®” (see, e.g. United States Patent Publication No: 2011-0206672; European Patent Publication No. EP 2371866, and; PCT Publication Nos. WO 1999/057150, WO 2003/025018, and WO 2013/013700). BiTEs are formed from a single polypeptide chain comprising tandem linked scFvs, while TandAbs are formed by the homo-dimerization of two identical polypeptide chains, each possessing a VH1, VL2, VH2, and VL2 Domain. The present invention provides bispecific binding molecules that are capable of enhancing an immune response directed to the killing of a target cell (e.g., a cancer cell or a pathogen-infected cell, a pathogen, etc.) expressing a Disease Antigen. Such bispecific binding molecules are capable of binding a “First Epitope” and a “Second Epitope,” wherein one of such epitopes is an epitope of CD16 and the other of such epitopes is an epitope of a Disease Antigen (“DA”). It is irrelevant whether a particular epitope is designated as the first vs. the Second Epitope; such notation having relevance only with respect to the presence and orientation of the domains of the polypeptide chains of the binding molecules of the present invention. Thus, the bispecific molecules of the present invention comprise “VLCD16”/“VHCD16” Domains that are capable of binding an epitope of CD16, and “VLDA”/“VHDA” Domains that are capable of binding an epitope of a Disease Antigen. The instant invention particular encompasses bispecific diabodies, BiTEs, antibodies, and TandAbs produced using any of the methods provided herein. In one embodiment, the CD16 Binding Molecules of the present invention will be bispecific diabodies and will comprise domains capable of binding both a first and a Second Epitope, but will lack an Fc Domain, and thus will be unable to bind FcγR molecules via an Fc-FcγR interaction. Such molecules are, however, able to bind to CD16 via the SDRs or CDRs of their CD16 Binding Domains. The absence of Fc domains thus serves to prevent the molecules from binding to non-CD16 FcγRs, such as the inhibitory receptor CD32B. The first polypeptide chain of such an embodiment of bispecific diabodies preferably comprises, in the N-terminal to C-terminal direction: an N-terminus, the VL Domain of a monoclonal antibody capable of binding either the First or Second Epitope (i.e., either VLCD16or VLDA), a first intervening spacer peptide (Linker 1), a VH Domain of a monoclonal antibody capable of binding the epitope of the Disease Antigen (if such first polypeptide chain contains VLCD16) or a VH Domain of a monoclonal antibody capable of binding CD16 (if such first polypeptide chain contains VLDA), a second intervening spacer peptide (Linker 2) optionally containing a cysteine residue, a Heterodimer-Promoting Domain and a C-terminus ( The second polypeptide chain of this embodiment of bispecific diabodies comprises, in the N-terminal to C-terminal direction: an N-terminus, the VL Domain of a monoclonal antibody capable of binding the First or Second Epitope (i.e., VLCD16or VLDA, and being the VL Domain not selected for inclusion in the first polypeptide chain of the diabody), an intervening spacer peptide (Linker 1), a VH Domain of a monoclonal antibody capable of binding either the First or Second Epitope (i.e., VHCD16or VHDA, and being the VH Domain not selected for inclusion in the first polypeptide chain of the diabody), a second intervening spacer peptide (Linker 2) optionally containing a cysteine residue, a Heterodimer-Promoting Domain and a C-terminus ( The VL Domain of the first polypeptide chain interacts with the VH Domain of the second polypeptide chain to form a first functional Epitope Binding Domain that is specific for one of the epitopes (e.g., the First Epitope). Likewise, the VL Domain of the second polypeptide chain interacts with the VH Domain of the first polypeptide chain in order to form a second functional Epitope Binding Domain that is specific for the other epitope (i.e., the Second Epitope). Thus, the selection of the VL and VH Domains of the first and second polypeptide chains is “coordinated,” such that the two polypeptide chains of the diabody collectively comprise VL and VH Domains capable of binding both the First Epitope and the Second Epitope (i.e., they collectively comprise VLCD16/VHCD16and VLDA/VHDA). Most preferably, the length of the intervening spacer peptide (i.e., “Linker 1,” which separates such VL and VH Domains) is selected to substantially or completely prevent the VL and VH Domains of the polypeptide chain from binding one another (for example consisting of from 0, 1, 2, 3, 4, 5, 6, 7, 8 or 9 intervening linker amino acid residues). Thus, the VL and VH Domains of the first polypeptide chain are substantially or completely incapable of binding one another. Likewise, the VL and VH Domains of the second polypeptide chain are substantially or completely incapable of binding one another. A preferred intervening spacer peptide (Linker 1) has the sequence (SEQ ID NO:16): The length and composition of the second intervening spacer peptide (“Linker 2”) is selected based on the choice of one or more polypeptide domains that promote such dimerization (i.e., a “Heterodimer-Promoting Domain”). Typically, the second intervening spacer peptide (Linker 2) will comprise 3-20 amino acid residues. In particular, where the employed Heterodimer-Promoting Domain(s) do/does not comprise a cysteine residue a cysteine-containing second intervening spacer peptide (Linker 2) is utilized. A cysteine-containing second intervening spacer peptide (Linker 2) will contain 1, 2, 3 or more cysteines. A preferred cysteine-containing spacer peptide (Linker 2) has the sequence GGCGGG (SEQ ID NO:17). Alternatively, Linker 2 does not comprise a cysteine (e.g., GGG, GGGS (SEQ ID NO:18), LGGGSG (SEQ ID NO:19), GGGSGGGSGGG (SEQ ID NO:20), AS TKG (SEQ ID NO:21), LEPKSS (SEQ ID NO:22), APSSS (SEQ ID NO:23), etc.) and a cysteine-containing Heterodimer-Promoting Domain, as described below is used. Optionally, both a cysteine-containing Linker 2 and a cysteine-containing Heterodimer-Promoting Domain are used. The Heterodimer-Promoting Domains may be GVEPKSC (SEQ ID NO:24) or VEPKSC (SEQ ID NO:25) or AEPKSC (SEQ ID NO:26) on one polypeptide chain and GFNRGEC (SEQ ID NO:27) or FNRGEC (SEQ ID NO:28) on the other polypeptide chain (US2007/0004909). In a preferred embodiment, the Heterodimer-Promoting Domains will comprise tandemly repeated coil domains of opposing charge for example, an “E-coil” Heterodimer-Promoting Domain (SEQ ID NO:29: EVAALEK-EVAALEK-EVAALEK-EVAALEK), whose glutamate residues will form a negative charge at pH 7, or a “K-coil” Heterodimer-Promoting Domain (SEQ ID NO:30: KVAALKE-KVAALKE-KVAALKE KVAALKE), whose lysine residues will form a positive charge at pH 7. The presence of such charged domains promotes association between the first and second polypeptides, and thus fosters heterodimer formation. Heterodimer-Promoting Domains that comprise modifications of the above-described E-coil and K-coil sequences so as to include one or more cysteine residues may be utilized. The presence of such cysteine residues permits the coil present on one polypeptide chain to become covalently bonded to a complementary coil present on another polypeptide chain, thereby covalently bonding the polypeptide chains to one another and increasing the stability of the diabody. Examples of such particularly preferred are Heterodimer-Promoting Domains include a Modified E-Coil having the amino acid sequence EVAACEK-EVAALEK-EVAALEK-EVAALEK (SEQ ID NO:31), and a modified K-coil having the amino acid sequence KVAACKE-KVAALKE-KVAALKE-KVAALKE (SEQ ID NO:32). As disclosed in WO 2012/018687, in order to improve the in vivo pharmacokinetic properties of diabodies, a diabody may be modified to contain a polypeptide portion of a serum-binding protein at one or more of the termini of the diabody. Most preferably, such polypeptide portion of a serum-binding protein will be installed at the C-terminus of a polypeptide chain of the diabody. Albumin is the most abundant protein in plasma and has a half-life of 19 days in humans. Albumin possesses several small molecule binding domains that permit it to non-covalently bind other proteins and thereby extend their serum half-lives. The Albumin-Binding Domain 3 (ABD3) of protein G of As disclosed in WO 2012/162068 (herein incorporated by reference), “deimmunized” variants of SEQ ID NO:33 have the ability to attenuate or eliminate MHC class II binding. Based on combinational mutation results, the following combinations of substitutions are considered to be preferred substitutions for forming such a deimmunized ABD: 66D/70S+71A; 66S/70S+71A; 66S/70S+79A; 64A/65A/71A; 64A/65A/71A+66S; 64A/65A/71A+66D; 64A/65A/71A+66E; 64A/65A/79A+66S; 64A/65A/79A+66D; 64A/65A/79A+66E. Variant ABDs having the modifications L64A, I65A and D79A or the modifications N66S, T7OS and D79A. Variant deimmunized ABD having the amino acid sequence: One embodiment of the present invention relates to multi-specific diabodies (e.g., bispecific, trispecific, tetraspecific, etc.) that comprise an Fc Domain and that are capable of simultaneously binding an epitope of CD16 and an epitope of a Disease Antigen. The Fc Domain of such molecules may be of any isotype (e.g., IgG1, IgG2, IgG3, or IgG4). The molecules may further comprise a CH1 Domain and/or a Hinge Domain. When present, the CH1 Domain and/or Hinge Domain may be of any isotype (e.g., IgG1, IgG2, IgG3, or IgG4), and is preferably of the same isotype as the desired Fc Domain. The addition of an IgG CH2-CH3 Domain to one or both of the diabody polypeptide chains, such that the complexing of the diabody chains results in the formation of an Fc Domain, increases the biological half-life and/or alters the valency of the diabody. Such diabodies comprise, two or more polypeptide chains whose sequences permit the polypeptide chains to covalently bind each other to form a covalently associated diabody that is capable of simultaneously binding the First Epitope and the Second Epitope. Incorporating an IgG CH2-CH3 Domains onto both of the diabody polypeptides will permit a two-chain bispecific Fc Domain-containing diabody to form ( Alternatively, incorporating IgG CH2-CH3 Domains onto only one of the diabody polypeptides will permit a more complex four-chain bispecific Fc Domain-containing diabody to form ( Fc Domain-containing diabody molecules of the present invention may include additional intervening spacer peptides (Linkers), generally such Linkers will be incorporated between a Heterodimer-Promoting Domain (e.g., an E-coil or K-coil) and a CH2-CH3 Domain and/or between a CH2-CH3 Domain and a Variable Domain (i.e., VH or VL). Typically, the additional Linkers will comprise 3-20 amino acid residues and may optionally contain all or a portion of an IgG Hinge Domain (preferably a cysteine-containing portion of an IgG Hinge Domain possessing 1, 2, 3 or more cysteine residues). Linkers that may be employed in the bispecific Fc Domain-containing diabody molecules of the present invention include: GGGS (SEQ ID NO:18), LGGGSG (SEQ ID NO:19), GGGSGGGSGGG (SEQ ID NO:20), AS TKG (SEQ ID NO:21), LEPKSS (SEQ ID NO:22), APSSS (SEQ ID NO:23), APSSSPME (SEQ ID NO:37), VEPKSADKTHTCPPCP (SEQ ID NO:38), LEPKSADKTHTCPPCP (SEQ ID NO:39), DKTHTCPPCP (SEQ ID NO:40), the scFv linker: GGGGSGGGGSGGGGS (SEQ ID NO:41); the “long” linker: GGGGSGGGSGGG (SEQ ID NO:42), GGC, and GGG. LEPKSS (SEQ ID NO:22) may be used in lieu of GGG or GGC for ease of cloning. Additionally, the amino acids GGG, or LEPKSS (SEQ ID NO:22) may be immediately followed by DKTHTCPPCP (SEQ ID NO:40) to form the alternate linkers: GGGDKTHTCPPCP (SEQ ID NO:43); and LEPKSSDKTHTCPPCP (SEQ ID NO:44). Bispecific Fc Domain-containing molecules of the present invention may incorporate an IgG Hinge Domain in addition to or in place of a linker. Exemplary Hinge Domains include: EPKSCDKTHTCPPCP (SEQ ID NO:5) from IgG1, ERKCCVECPPCP (SEQ ID NO:6) from IgG2, ELKTPLGDTTHTCPRCPEPKSCDTPPPCPRCPEPKSCDTPPPCPRCPEPKS CDTPPPCPRCP (SEQ ID NO:7) from IgG3, ESKYGPPCPSCP (SEQ ID NO:8) from IgG4, and ESKYGPPCPPCP (SEQ ID NO:9) an IgG4 Hinge variant comprising a stabilizing S228P substitution (as numbered by the EU index as set forth in Kabat) to reduce strand exchange. As provided in In a specific embodiment, diabodies of the present invention are bispecific, tetravalent (i.e., possess four Epitope Binding Domains), Fc-containing diabodies that are composed of four total polypeptide chains ( In a further embodiment, the Fc Domain-containing diabodies of the present invention may comprise three polypeptide chains. The first polypeptide of such a diabody contains three domains: (i) a VL1-containing Domain, (ii) a VH2-containing Domain and (iii) a Domain containing a CH2-CH3 sequence. The second polypeptide of such a diabody contains: (i) a VL2-containing Domain, (ii) a VH1-containing Domain and (iii) a Domain that promotes heterodimerization and covalent bonding with the diabody's first polypeptide chain. The third polypeptide of such a diabody comprises a CH2-CH3 sequence. Thus, the first and second polypeptide chains of such a diabody associate together to form a VL1/VH1 Epitope Binding Domain that is capable of binding either the First or Second Epitope, as well as a VL2/VH2 Epitope Binding Domain that is capable of binding the other of such epitopes. The first and second polypeptides are bonded to one another through a disulfide bond involving cysteine residues in their respective Third Domains. Notably, the first and third polypeptide chains complex with one another to form an Fc Domain that is stabilized via a disulfide bond. Such bispecific diabodies have enhanced potency. In a specific embodiment, diabodies of the present invention are bispecific, bivalent (i.e., possess two Epitope Binding Domains), Fc-containing diabodies that are composed of three total polypeptide chains ( In a further embodiment, the Fc Domain-containing diabodies may comprise a total of five polypeptide chains. In a particular embodiment, two of the five polypeptide chains have the same amino acid sequence. The first polypeptide chain of such a diabody contains: (i) a VH1-containing Domain, (ii) a CH1-containing Domain, and (iii) a Domain containing a CH2-CH3 sequence. The first polypeptide chain may be the Heavy Chain of an antibody that contains a VH1 and a Heavy Chain constant region. The second and fifth polypeptide chains of such a diabody contain: (i) a VL1-containing Domain, and (ii) a CL-containing Domain. The second and/or fifth polypeptide chains of such a diabody may be Light Chains of an antibody that contains a VL1 complementary to the VH1 of the first/third polypeptide chain. The first, second and/or fifth polypeptide chains may be isolated from a naturally occurring antibody. Alternatively, they may be constructed recombinantly. The third polypeptide chain of such a diabody contains: (i) a VH1-containing Domain, (ii) a CH1-containing Domain, (iii) a Domain containing a CH2-CH3 sequence, (iv) a VL2-containing Domain, (v) a VH3-containing Domain and (vi) a Heterodimer-Promoting Domain, where the Heterodimer-Promoting Domains promote the dimerization of the third chain with the fourth chain. The fourth polypeptide of such diabodies contains: (i) a VL3-containing Domain, (ii) a VH2-containing Domain and (iii) a Domain that promotes heterodimerization and covalent bonding with the diabody's third polypeptide chain. Thus, the first and second, and the third and fifth, polypeptide chains of such diabodies associate together to form two VL1/VH1 Epitope Binding Domains capable of binding a First Epitope. The third and fourth polypeptide chains of such diabodies associate together to form a VL2/VH2 Epitope Binding Domain that is capable of binding a Second Epitope, as well as a VL3/VH3 Epitope Binding Domain that is capable of binding a Third Epitope. The first and third polypeptides are bonded to one another through a disulfide bond involving cysteine residues in their respective constant regions. Notably, the first and third polypeptide chains complex with one another to form an Fc Domain. Such multispecific diabodies have enhanced potency. The VL and VH Domains of the polypeptide chains are selected so as to form VL/VH Epitope Binding Domains specific for a desired epitope. The VL/VH Epitope Binding Domains formed by the association of the polypeptide chains may be the same or different so as to permit tetravalent binding that is mono-specific, bispecific, trispecific or tetraspecific. In particular, the VL and VH Domains maybe selected such that a multivalent diabody may comprise two Binding Domains for a First Epitope and two Binding Domains for a Second Epitope, or three Binding Domains for a First Epitope and one Binding Domain for a Second Epitope, or two Binding Domains for a First Epitope, one Binding Domain for a Second Epitope and one Binding Domain for a Third Epitope (as depicted in In a specific embodiment, diabodies of the present invention are bispecific, tetravalent (i.e., possess four Epitope Binding Domains), Fc-containing diabodies that are composed of five total polypeptide chains having two Epitope Binding Domains immunospecific for the First Epitope, and two Epitope Binding Domains specific for the Second Epitope. In another embodiment, the bispecific, tetravalent, Fc-containing diabodies of the invention comprise three Epitope Binding Domains immunospecific for the First Epitope and one Epitope Binding Domain specific for the Second Epitope. As provided above, the VL and VH Domains may be selected to permit trispecific binding. Accordingly, the invention also encompasses trispecific, tetravalent, Fc-containing diabodies. The trispecific, tetravalent, Fc-containing diabodies of the invention comprise two Epitope Binding Domains immunospecific for the First Epitope, one Epitope Binding Domain immunospecific for the Second Epitope, and one Epitope Binding Domain immunospecific for the Third Epitope. In traditional immune function, the interaction of antibody-antigen complexes with cells of the immune system results in a wide array of responses, ranging from effector functions such as antibody-dependent cytotoxicity, mast cell degranulation, and phagocytosis to immunomodulatory signals such as regulating lymphocyte proliferation and antibody secretion. All of these interactions are initiated through the binding of the Fc Domain of antibodies or immune complexes to specialized cell surface receptors on hematopoietic cells. As discussed above, the diversity of cellular responses triggered by antibodies and immune complexes results from the structural heterogeneity of the three Fc receptors: FcγRT (CD64), FcγRII (CD32), and FcγRIII (CD16). FcγRT (CD64), FcγRIIA (CD32A) and FcγRIII (CD16) are activating (i.e., immune system enhancing) receptors; FcγRIIB (CD32B) is an inhibiting (i.e., immune system dampening) receptor. In addition, interaction with the neonatal Fc Receptor (FcRn) mediates the recycling of IgG molecules from the endosome to the cell surface and release into the blood. The amino acid sequence of exemplary wild-type IgG1 (SEQ ID NO:12), IgG2 (SEQ ID NO:13), IgG3 (SEQ ID NO:14), and IgG4 (SEQ ID NO:15) are presented above. Modification of the Fc Domain may lead to an altered phenotype, for example altered serum half-life, altered stability, altered susceptibility to cellular enzymes or altered effector function. It may therefore be desirable to modify an Fc Domain-containing binding molecule of the present invention with respect to effector function, for example, so as to enhance the effectiveness of such molecule in treating cancer. Reduction or elimination of Fc Domain-mediated effector function is desirable in certain cases, for example in the case of antibodies whose mechanism of action involves blocking or antagonism, but not killing of the cells bearing a target antigen. Increased effector function is generally desirable when directed to undesirable cells, such as tumor and foreign cells, where the FcγRs are expressed at low levels, for example, tumor-specific B cells with low levels of FcγRIIB (e.g., non-Hodgkin's lymphoma, CLL, and Burkitt's lymphoma). Molecules of the invention possessing such conferred or altered effector function activity are useful for the treatment and/or prevention of a disease, disorder or infection in which an enhanced efficacy of effector function activity is desired. Accordingly, in certain embodiments, the Fc Domain of the Fc Domain-containing molecules of the present invention may be an engineered variant Fc Domain. Although the Fc Domain of the bispecific Fc Domain-containing molecules of the present invention may possess the ability to bind one or more Fc receptors (e.g., FcγR(s)), more preferably such variant Fc Domain have altered binding FcγRIA (CD64), FcγRIIA (CD32A), FcγRIIB (CD32B), FcγRIIIA (CD16a) or FcγRIIIB (CD16b) (relative to the binding exhibited by a wild-type Fc Domain), e.g., will have enhanced binding an activating receptor and/or will have substantially reduced or no ability to bind inhibitory receptor(s). Thus, the Fc Domain of the Fc Domain-containing molecules of the present invention may include some or all of the CH2 Domain and/or some or all of the CH3 Domain of a complete Fc Domain, or may comprise a variant CH2 and/or a variant CH3 sequence (that may include, for example, one or more insertions and/or one or more deletions with respect to the CH2 or CH3 domains of a complete Fc Domain). Such Fc Domains may comprise non-Fc polypeptide portions, or may comprise portions of non-naturally complete Fc Domains, or may comprise non-naturally occurring orientations of CH2 and/or CH3 Domains (such as, for example, two CH2 Domains or two CH3 Domains, or in the N-terminal to C-terminal direction, a CH3 Domain linked to a CH2 Domain, etc.). Fc Domain modifications identified as altering effector function are known in the art, including modifications that increase binding activating receptors (e.g., FcγRIIA (CD16A) and reduce binding inhibitory receptors (e.g., FcγRIIB (CD32B) (see, e.g., Stavenhagen, J. B. et al. (2007) “ Exemplary variants of human IgG1 Fc Domains with reduced binding CD32B and/or increased binding CD16A contain F243L, R292P, Y300L, V305I or P396L substitutions, wherein the numbering is that of the EU index as in Kabat. These amino acid substitutions may be present in a human IgG1 Fc Domain in any combination. In one embodiment, the variant human IgG1 Fc Domain contains a F243L, R292P and Y300L substitution. In another embodiment, the variant human IgG1 Fc Domain contains a F243L, R292P, Y300L, V305I and P396L substitution. In certain embodiments, it is preferred for the Fc Domains of the Fc Domain-containing binding molecules of the present invention to exhibit decreased (or substantially no) binding FcγRIA (CD64), FcγRIIA (CD32A), FcγRIIB (CD32B), FcγRIIIA (CD16a) or FcγRIIIB (CD16b) (relative to the binding exhibited by the wild-type IgG1 Fc Domain (SEQ ID NO:12). In a specific embodiment, the Fc Domain-containing binding molecules of the present invention comprise an IgG Fc Domain that exhibits reduced antibody-dependent cell-mediated cytotoxicity (ADCC) effector function. In a preferred embodiment, the CH2-CH3 Domains of such binding molecules include any 1, 2, 3, or 4 of the substitutions: L234A, L235A, D265A, N297Q, and N297G, wherein the numbering is that of the EU index as in Kabat. In another embodiment, the CH2-CH3 Domains contain an N297Q substitution, an N297G substitution, L234A and L235A substitutions or a D265A substitution, as these mutations abolish FcR binding. Alternatively, a CH2-CH3 Domain of a naturally occurring Fc Domain that inherently exhibits decreased (or substantially no) binding FcγRIIIA (CD16a) and/or reduced effector function (relative to the binding and effector function exhibited by the wild-type IgG1 Fc Domain (SEQ ID NO:12)) is utilized. In a specific embodiment, the Fc Domain-containing binding molecules of the present invention comprise an IgG2 Fc Domain (SEQ ID NO:13), an IgG3 Fc Domain (SEQ ID NO:14) or an IgG4 Fc Domain (SEQ ID NO:15). When an IgG4 Fc Domain is utilized, the instant invention also encompasses the introduction of a stabilizing mutation, such as the Hinge Region S228P substitution described above (see, e.g., SEQ ID NO:11). Since the N297G, N297Q, L234A, L235A and D265A substitutions abolish effector function, in circumstances in which effector function is desired, these substitutions would preferably not be employed. A preferred IgG1 sequence for the CH2 and CH3 Domains of the Fc Domain-containing molecules of the present invention having reduced or abolished effector function will comprise the substitutions L234A/L235A (SEQ ID NO:45): wherein X is lysine (K) or is absent. A second preferred IgG1 sequence for the CH2 and CH3 Domains of the Fc Region-containing molecules of the present invention comprises an S442C substitution (shown underlined), so as to permit two CH3 domains to be covalently bonded to one another via a disulfide bond or to permit conjugation of a drug moiety. The amino acid sequence of such molecule is (SEQ ID NO:46): A third preferred IgG1 sequence for the CH2 and CH3 Domains of the Fc Region-containing molecules of the present invention comprises the L234A/L235A substitutions (shown underlined) that reduce or abolish effector function and the S442C substitution (shown underlined) that permits two CH3 domains to be covalently bonded to one another via a disulfide bond or conjugation of a drug moiety. The amino acid sequence of such molecule is (SEQ ID NO:47): The serum half-life of proteins comprising Fc Domains may be increased by increasing the binding affinity of the Fc Domain for FcRn. The term “half-life” as used herein means a pharmacokinetic property of a molecule that is a measure of the mean survival time of the molecules following their administration. Half-life can be expressed as the time required to eliminate fifty percent (50%) of a known quantity of the molecule from a subject's body (e.g., a human patient or other mammal) or a specific compartment thereof, for example, as measured in serum, i.e., circulating half-life, or in other tissues. In general, an increase in half-life results in an increase in mean residence time (MRT) in circulation for the molecule administered. In some embodiments, the Fc Domain-containing binding molecules of the present invention comprise a variant Fc Domain that comprises at least one amino acid modification relative to a wild-type Fc Domain, such that the molecule has an increased half-life (relative to such molecule if comprising a wild-type Fc Domain). In some embodiments, the Fc Domain-containing binding molecules of the present invention comprise a variant IgG Fc Domain that comprises a half-life extending amino acid substitution at one or more positions selected from the group consisting of 238, 250, 252, 254, 256, 257, 256, 265, 272, 286, 288, 303, 305, 307, 308, 309, 311, 312, 317, 340, 356, 360, 362, 376, 378, 380, 382, 413, 424, 428, 433, 434, 435, and 436, wherein the numbering is that of the EU index as in Kabat. Numerous mutations capable of increasing the half-life of an Fc Domain-containing molecule are known in the art and include, for example M252Y, S254T, T256E, and combinations thereof. For example, see the mutations described in U.S. Pat. Nos. 6,277,375, 7,083,784; 7,217,797, 8,088,376; U.S. Publication Nos. 2002/0147311; 2007/0148164; and PCT Publication Nos. WO 98/23289; WO 2009/058492; and WO 2010/033279, which are herein incorporated by reference in their entireties. In some embodiments, the Fc Domain-containing binding molecules of the present invention exhibiting enhanced half-life possess a variant Fc Domain comprising substitutions at two or more of Fc Domain residues 250, 252, 254, 256, 257, 288, 307, 308, 309, 311, 378, 428, 433, 434, 435 and 436. In particular, two or more substitutions selected from: T250Q, M252Y, S254T, T256E, K288D, T307Q, V308P, A378V, M428L, N434A, H435K, and Y436I, wherein the numbering is that of the EU index as in Kabat. In a specific embodiment, such molecules may possess a variant IgG Fc Domain comprising the substitution:
In a preferred embodiment, an Fc Domain-containing CD16 x DA Binding Molecule of the present invention possesses a variant IgG Fc Region comprising any 1, 2, or 3 of the substitutions: M252Y, S254T and T256E. The invention further encompasses CD16 x DA Binding Molecules possessing variant Fc Regions comprising:
An IgG1 sequence for the CH2 and CH3 Domains of the Fc Domain-containing molecules of the present invention that provides an increased half-life (and that has a 10-fold increase in binding to both cynomolgus monkey and human FcRn) (Dall'Acqua, W. F. et al. (2006) “ wherein X is lysine (K) or is absent. An alternative IgG1 sequence for the CH2 and CH3 Domains of the Fc Domain-containing molecules of the present invention combining the reduced or abolished effector function provided by the substitutions L234A/L235A and the increased serum half-life provided by the substitutions M252Y/S254T/T256E is provided by SEQ ID NO: 49: wherein X is lysine (K) or is absent. A further preferred IgG1 sequence for the CH2 and CH3 Domains of the Fc Region-containing molecules of the present invention comprises the L234A/L235A substitutions (shown underlined) that reduce or abolish effector function and the M252Y, S254T and T256E substitutions (shown underlined), so as to extend the serum half-life and the S442C substitution (shown underlined), so as to permit two CH3 domains to be covalently bonded to one another via a disulfide bond or to permit conjugation of a drug moiety. The amino acid sequence of such molecule is (SEQ ID NO:50): For certain antibodies, diabodies and trivalent binding molecules that are desired to have Fc-Domain-containing polypeptide chains of differing amino acid sequence (e.g., whose Fc Domain-containing polypeptide chains are desired to not be identical), it is desirable to reduce or prevent homodimerization from occurring between the CH2-CH3 Domains of identical chains (e.g., two first polypeptide chains or between the CH2-CH3 Domains of two third polypeptide chains). The CH2 and/or CH3 Domains of such polypeptide chains need not be identical in sequence, and advantageously are modified to foster heterodimer complexing between the two polypeptide chains. For example, an amino acid substitution (preferably a substitution with an amino acid comprising a bulky side group forming a “knob”, e.g., tryptophan) can be introduced into the CH2 or CH3 Domain such that steric interference will prevent interaction with a similarly mutated domain and will obligate the mutated domain to pair with a domain into which a complementary, or accommodating mutation has been engineered, i.e., “the hole” (e.g., a substitution with glycine). Such sets of mutations can be engineered into any pair of polypeptides comprising CH2-CH3 Domains that forms an Fc Domain to foster heterodimerization. Methods of protein engineering to favor heterodimerization over homodimerization are well-known in the art, in particular with respect to the engineering of immunoglobulin-like molecules, and are encompassed herein (see e.g., Ridgway et al. (1996) “‘ A preferred knob is created by modifying an IgG Fc Domain to contain the modification T366W. A preferred hole is created by modifying an IgG Fc Domain to contain the modification T366S, L368A and Y407V. To aid in purifying a hole-bearing polypeptide chain homodimer from the final bispecific heterodimeric Fc Domain-containing molecule, the Protein A Binding Domain of the hole-bearing CH2 and CH3 Domains of a polypeptide chain is preferably mutated by amino acid substitution at position 435 (H435R). Thus, the hole-bearing polypeptide chain homodimer will not bind protein A, whereas the bispecific heterodimer will retain its ability to bind protein A via the Protein A Binding Domain. In an alternative embodiment, the hole-bearing polypeptide chain may incorporate amino acid substitutions at positions 434 and 435 (N434A/N435K). A preferred IgG1 amino acid sequence for the CH2 and CH3 Domains of one Fc Domain-containing polypeptide chain of an Fc Domain-containing molecule of the present invention will have the “knob-bearing” sequence (SEQ ID NO:51): wherein X is lysine (K) or is absent. An alternative IgG1 amino acid sequence for the CH2 and CH3 Domains of one Fc Domain-containing polypeptide chain of an Fc Domain-containing molecule of the present invention having a M252Y/S254T/T256E substitution and a “knob-bearing” sequence is SEQ ID NO:52: wherein X is lysine (K) or is absent. A preferred IgG1 amino acid sequence for the CH2 and CH3 Domains of the other Fc Domain-containing polypeptide chain of an Fc Domain-containing molecule of the present invention having two polypeptide chains (or the third polypeptide chain of an Fc Domain-containing molecule having three, four, or five polypeptide chains) will have the “hole-bearing” sequence (SEQ ID NO:53): wherein X is lysine (K) or is absent. An alternative IgG1 amino acid sequence for the CH2 and CH3 Domains of the other Fc Domain-containing polypeptide chain of an Fc Domain-containing molecule of the present invention having a M252Y/S254T/T256E substitution and a “hole-bearing” sequence is SEQ ID NO:54: wherein X is lysine (K) or is absent. As will be noted, the CH2-CH3 Domains of SEQ ID NO:51, SEQ ID NO:52, SEQ ID NO:53, and SEQ ID NO:54 include a substitution at position 234 with alanine and 235 with alanine, and thus form an Fc Domain exhibit decreased (or substantially no) binding FcγRIA (CD64), FcγRIIA (CD32A), FcγRIIB (CD32B), FcγRIIIA (CD16a) or FcγRIIIB (CD16b) (relative to the binding exhibited by the wild-type Fc Domain (SEQ ID NO:12). The invention also encompasses such CH2-CH3 Domains, which comprise the wild-type alanine residues, alternative and/or additional substitutions which modify effector function and/or FγR binding activity of the Fc Domain. The invention also encompasses such CH2-CH3 Domains, which further comprise one or more half-live extending amino acid substitutions. In particular, the invention encompasses such hole-bearing and such knob-bearing CH2-CH3 Domains which further comprise the M252Y/S254T/T256E. An IgG4 amino acid sequence for the CH2 and CH3 Domains of the one Fc Domain-containing polypeptide chain of an Fc Domain-containing molecule of the present invention has enhanced serum half-life (relative to IgG1 CH2 and CH3 Domains) due to its possession of Y252/T254/E256 (SEQ ID NO:55): wherein X is lysine (K) or is absent. A “knob-bearing” variant of such an IgG4 CH2-CH3 amino acid sequence has the amino acid sequence of SEQ ID NO:56: wherein X is lysine (K) or is absent. A “hole-bearing” variant of such an IgG4 CH2-CH3 amino acid sequence has the amino acid sequence of SEQ ID NO:57: wherein X is lysine (K) or is absent. It is preferred that the first polypeptide chain will have a “knob-bearing” CH2-CH3 sequence, such as that of SEQ ID NO:51 or SEQ ID NO:52. However, as will be recognized, a “hole-bearing” CH2-CH3 Domain (e.g., SEQ ID NO:53 or SEQ ID NO:54) could be employed in the first polypeptide chain, in which case, a “knob-bearing” CH2-CH3 Domain (e.g., SEQ ID NO:51 or SEQ ID NO:52) would be employed in the second polypeptide chain of an Fc Domain-containing molecule of the present invention having two polypeptide chains (or in the third polypeptide chain of an Fc Domain-containing molecule having three, four, or five polypeptide chains). In other embodiments, the invention encompasses Fc Domain-containing binding molecules comprising CH2 and/or CH3 Domains that have been engineered to favor heterodimerization over homodimerization using mutations known in the art, such as those disclosed in PCT Publication No. WO 2007/110205; WO 2011/143545; WO 2012/058768; WO 2013/06867, all of which are incorporated herein by reference in their entirety. A further embodiment of the present invention relates to trivalent binding molecules comprising an Fc Domain capable of simultaneously binding a First Epitope, a Second Epitope and a Third Epitope, wherein at least one of such epitopes is not identical to another. Such trivalent binding molecules comprise three Epitope Binding Domains, two of which are Diabody-Type Binding Domains, which provide Binding Domain A and Binding Domain B, and one of which is a Fab-Type Binding Domain, or an scFv-Type Binding Domain, which provides Binding Domain C (see, e.g., Typically, the trivalent Binding Molecules of the present invention will comprise four different polypeptide chains (see In certain embodiments, the first polypeptide chain of such trivalent binding molecules of the present invention contains: (i) a VL1-containing Domain, (ii) a VH2-containing Domain, (iii) a Heterodimer-Promoting Domain, and (iv) a Domain containing a CH2-CH3 sequence. The VL1 and VL2 Domains are located N-terminal or C-terminal to the CH2-CH3-containing domain as presented in Table 4 (also see, The Light Chain Variable Domain of the first and second polypeptide chains are separated from the Heavy Chain Variable Domains of such polypeptide chains by an intervening spacer peptide having a length that is too short to permit their VL1/VH2 (or their VL2/VH1) domains to associate together to form an Epitope Binding Domain capable of binding either the First or Second Epitope. A preferred intervening spacer peptide (Linker 1) for this purpose has the sequence (SEQ ID NO:16): As described above, the trivalent binding molecules of the present invention may comprise three polypeptides. Trivalent binding molecules comprising three polypeptide chains may be obtained by linking the domains of the fourth polypeptide N-terminal to the VH3-containing Domain of the third polypeptide (e.g., using an intervening spacer peptide (Linker 4)). Alternatively, a third polypeptide chain of a trivalent binding molecule of the invention containing the following domains is utilized: (i) a VL3-containing Domain, (ii) a VH3-containing Domain, and (iii) a Domain containing a CH2-CH3 sequence, wherein the VL3 and VH3 are spaced apart from one another by an intervening spacer peptide that is sufficiently long (at least 9 or more amino acid residues) so as to allow the association of these domains to form an Epitope Binding Domain. One preferred intervening spacer peptide for this purpose has the sequence: It will be understood that the VL1/VH1, VL2/VH2, and VL3/VH3 Domains of such trivalent binding molecules may be different so as to permit binding that is mono-specific, bispecific or trispecific. In particular, the VL and VH Domains may be selected such that a trivalent binding molecule comprises two Binding Domains for a First Epitope and one Binding Domains for a Second Epitope, or one Binding Domain for a First Epitope and two Binding Domains for a Second Epitope, or one Binding Domain for a First Epitope, one Binding Domain for a Second Epitope and one Binding Domain for a Third Epitope. The general structure of the polypeptide chains of representative trivalent binding molecules of invention is provided in As provided above, such trivalent binding molecules may comprise three, four, five, or more polypeptide chains. As stated above, the present invention is directed to a CD16 x DA Binding Molecule (e.g., an antibody, a diabody, an scFv, an antibody, a TandAb, etc.) comprising a Binding Domain capable of binding an epitope of CD16 (i.e., a “CD16-Binding Domain”) and a Binding Domain capable of binding an epitope of a Disease Antigen (i.e., a “Disease Antigen-Binding Domain”). The invention thus encompasses binding molecules comprising one or more of the VH and/or VL Domains of an antibody that binds to CD16, or more preferably, the CDRH1, CDRH2, and CDRH3, and the CDRL1, CDRL2 and CDRL3 portions of such Domains. In a preferred embodiment of the invention, such binding molecules will additionally contain Binding Domains sufficient to permit such molecules to bind to epitopes of one, two, three or or more than three Disease Antigens. The present invention is also directed to pharmaceutical compositions that comprise such molecule(s). By possessing Binding Domains sufficient to immunospecifically bind CD16 and a Disease Antigen, the molecules of the present invention have the ability to co-localize CD16-expressing cells (and especially Natural Killer cells) to the site(s) of cells expressing the Disease Antigen so as to enhance the likelihood of ADCC-mediated killing of the target cell. As discussed above, such molecules may be bispecific, or may be capable of binding more than two epitopes. In one embodiment, such CD16-binding molecules of the present invention will be mono-specific so as to possess the ability to bind to only a single epitope of CD16 and only a single epitope of a Disease Antigen. Alternatively, such molecules may be multi-specific, i.e., capable of binding one, two, three or four total epitopes, which may be apportioned in any manner to bind one, two or three epitope(s) of CD16 (which two or three CD16 epitopes may be the same or different) and three, two or one epitope(s) of one or more Disease Antigen(s). Thus, where such molecules are capable of immunospecifically binding to only a single Disease Antigen, they may be capable of immunospecifically binding to only one CD16 epitope and to one, two or three epitope(s) of the Disease Antigen (which two Disease Antigen epitopes may be the same or different, and which three epitopes may be the same, or may be different, or may be two epitopes that are the same and one epitope that is different), or they may be capable of immunospecifically binding to only two CD16 epitopes (which two epitopes may be the same or different) and one or two epitope(s) of the Disease Antigen (which two Disease Antigen epitopes may be the same or different), or they may be capable of immunospecifically binding to three CD16 epitopes (which three epitopes may be the same, or may be different or may be two epitopes that are the same and one epitope that is different) and 1 epitope of the Disease Antigen. Similarly, where such molecules are capable of immunospecifically binding to two different Disease Antigens (e.g., a First Disease Antigen and a Second Disease Antigen), they may be capable of immunospecifically binding to only one CD16 epitope and to one or two epitope(s) of the First Disease Antigen (which two First Disease Antigen epitopes may be the same or different) and two or one epitope(s) of the Second Disease Antigen (which two Second Disease Antigen epitopes may be the same or different), or they may be capable of immunospecifically binding to only two CD16 epitopes (which two epitopes may be the same or different) and one epitope of the First Disease Antigen and one epitope of the Second Disease Antigen. Similarly, such molecules may be capable of immunospecifically binding to three different Disease Antigens (e.g., a First Disease Antigen, a Second Disease Antigen and a Third Disease Antigen) and only one CD16 epitope. Thus, for example, such molecules may bind:
Table 6 illustrates possible combination binding specificities of exemplary molecules of the invention. By forming more complex molecules, one may obtain CD16-binding molecules that are capable of binding one or more Disease Antigens and that possess more than four epitope binding domains. Thus, no limitation is placed on the nature of epitopes or additional epitopes that may be bound by the molecules of the present invention other than that such additional binding capability does not prevent the molecule or Binding Domain thereof that is capable of binding to an epitope of CD16 from such binding and does not prevent the molecule or Binding Domain thereof that is capable of binding to an epitope of a Disease Antigen from such binding. Thus, the CD16 Binding molecules of the present invention may possess Epitope Binding Domains alternative or additional Epitope Binding Domains. As an example, the invention contemplates a binding molecule that comprises a First Epitope Binding Domain capable of immunospecifically binding an epitope of CD16 and a Second Epitope Binding Domain that is capable of immunospecifically binding an epitope of a Disease Antigen that is arrayed on the surface of such target cell and a Third Epitope Binding Domain capable of immunospecifically binding a different cell surface molecule, such as a non-CD16 cell surface molecule of a Natural Killer cell. The present invention is directed molecules (e.g., an antibody, a diabody, an scFv, an antibody, a TandAb, etc.) capable of binding human CD16 by virtue of their possession of a CD16 Binding Domain. The present invention is particularly directed to such CD-16 Binding Molecules that are CD16 x DA Binding Molecules. Listed below are exemplary antibodies that may be used to produce the binding molecules and combination therapy of the present invention. The present invention provides murine anti-human CD16 monoclonal antibodies: CD16-M1 and CD16-M2, and their humanized derivatives: hCD16-M1 and hCD16-M2, which are novel, high affinity anti-human CD16 monoclonal antibodies that bind well to both the CD16 158F allotype and the CD16 158V allotype, and that bind CD16 at a site that does not block CD16-IgG binding. Such antibodies are particularly preferred for the purposes of the present invention since they can be readily employed in a patient population irrespective of its 158F/158V CD16A polymorphisms. The amino acid sequence of the VH Domain of murine anti-human CD16 monoclonal antibody CD16-M1 (SEQ ID NO:64) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of murine anti-human CD16 monoclonal antibody CD16-M1 (SEQ ID NO:65) is shown below (CDRLresidues are shown underlined): The CDRs of anti-human CD16 monoclonal antibody CD16-M1 are shown in Table 7. hCD16-M1 is a humanized derivative of murine anti-human CD16 monoclonal antibody CD16-M1. The amino acid sequence of the VH Domain of hCD16-M1 (SEQ ID NO:72) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of hCD16-M1 (SEQ ID NO:73) is shown below (CDRLresidues are shown underlined): hCD16-M1A is an optimized derivative of the humanized anti-human CD16 monoclonal antibody hCD16-M1. hCD16-M1A comprises the VL Domain of hCD16-M1 (SEQ ID NO:73) and an optimized VH Domain comprising mutations in CDRH3. The amino acid sequence of the optimized VH Domain of hCD16-M1A (SEQ ID NO:58) is shown below (the mutated CDRH3 residues are shown underlined): hCD16-M1B is an optimized derivative of the humanized anti-human CD16 monoclonal antibody hCD16-M1. hCD16-M1B comprises the VH Domain of hCD16-M1 (SEQ ID NO:72) and an optimized VL Domain comprising mutations in CDRL3. The amino acid sequence of the optimized VL Domain of hCD16-M1B (SEQ ID NO:59) is shown below (the mutated CDRL3 residues are shown underlined): hCD16-M1AB is an optimized derivative of the humanized anti-human CD16 monoclonal antibody hCD16-M1. hCD16-M1AB comprises the optimized VH Domain of hCD16-M1A (SEQ ID NO:58) and the optimized VL Domain of hCD16-M1B (SEQ ID NO:59). The CDRs of humanized anti-human CD16 monoclonal antibody hCD16-M1 and the optimized anti-human CD16 monoclonal antibodies hCD16-M1A, hCD16-M1B, hCD16-M1AB are shown in Table 8. As will be recognized, CDRL1 of hCD16-M1, hCD16-M1A, hCD16-M1B, and hCD16-M1AB (RASQNVGTHVA; SEQ ID NO:74) differs from CDRL1 of CD16-M1 (KASQNVGTHVA; SEQ ID NO:69) in its first residue. Either CDRL1 may be employed interchangeably, and the present invention encompasses humanized, and optimized CD16 Binding Molecules that comprise a CD16 Epitope Binding Domain having the amino acid sequence of 1, 2 or 3 of the following CDRHs, and/or 1, 2 or 3 of the such CDRLs. The amino acid sequence of the VH Domain of murine anti-human CD16 monoclonal antibody CD16-M2 (SEQ ID NO:75) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of murine anti-human CD16 monoclonal antibody CD16-M2 (SEQ ID NO:76) is shown below (CDRLresidues are shown underlined): The CDRs of anti-human CD16 monoclonal antibody CD16-M2 are shown in Table 9: hCD16-M2 is a humanized derivative of murine anti-human CD16 monoclonal antibody CD16-M2. Humanization resulted in two suitable VH Domains (hCD16-M2 VH1 and hCD16-M2 VH2), either of which may be employed with the obtained humanized VL Domain (hCD16-M2 VL1). The amino acid sequence of VH Domain hCD16-M2 VH1 (SEQ ID NO:83) is shown below (CDRHresidues are shown underlined): The amino acid sequence of VH Domain hCD16-M2 VH2 (SEQ ID NO:84) is shown below (CDRHresidues are shown underlined): As will be recognized, the amino acid sequence of hCD16-M2 VH1 (SEQ ID NO:83) differs from that of hCD16-M2 VH2 (SEQ ID NO:84) in possessing a T98R substitution in the residue that immediately precedes CDRH3 (shown boxed above). The amino acid sequence of the VL Domain hCD16-M2 VL1 (SEQ ID NO:85) is shown below (CDRLresidues are shown underlined): The CDRs of humanized anti-human CD16 monoclonal antibody hCD16-M2 are shown in Table 10. The CD16 x DA Binding Molecules of the present invention, and particularly the trispecific CD16 x DA Binding Molecules of the present invention may comprise a binding site for a non-CD16 cell surface molecule of an effector cell. As used herein, the term “effector cell” denotes a cell that directly or indirectly mediates the killing of target cells (e.g., foreign cells, infected cells or cancer cells). Examples of effector cells include helper T Cells, cytotoxic T Cells, Natural Killer (NK) cells, plasma cells (antibody-secreting B cells), macrophages and granulocytes. Preferred cell surface molecules of such cells include CD2, CD3, CD8, CD16, TCR, and the NKG2D receptor. Accordingly, molecules capable of immunospecifically binding an epitope of such molecules, or to other effector cell surface molecules may be used in accordance with the principles of the present invention. Exemplary antibodies, whose VH and VL Domains may be used to construct molecules capable of mediating the redirected killing of a target cell are provided below. A preferred non-CD16 cell surface molecule of a Natural Killer effector cell is the NKG2D receptor. The NKG2D receptor is expressed on all human (and other mammalian) Natural Killer cells (Bauer, S. et al. (1999) “ The amino acid sequence of the VH Domain of KYK-1.0 (SEQ ID NO:86) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of KYK-1.0 (SEQ ID NO:87) is shown below (CDRLresidues are shown underlined): The amino acid sequence of a VH Domain of KYK-2.0 (SEQ ID NO:88) is shown below (CDRHresidues are shown underlined): The amino acid sequence of a VL Domain of KYK-2.0 (SEQ ID NO:89) is shown below (CDRLresidues are shown underlined): Other exemplary antibodies that bind to the cell surface of a Natural Killer cell include antibodies: A1, AC2, EPR3678(2), EPR20461, EPR20627 and IMG17B5F11 (which bind CD39); TB01, HNK-1/Leu-7 and NK1 (which bind CD57); FN50 (which binds CD69); 5B5, B-L2, TS82b and C33 (which bind CD82); 3B3, B199.2 and EP7169 (which bind CD161); 17D9 (which binds CLEC1B); 2F9 (which binds KIR2DL1); EPR8825 (which binds KIR2DL2); mAb 33 (which binds KIR2DL4); 11E3, 17B4, EPR4392(2), EPR20261 and EPR 20627 (which bind Lymphocyte Activation Gene 3); A10, C7, CX5, 1D11 and MM0489-10R27 (which bind NKG2D); BMK13 (which binds PRG2); EPR9916 (which binds SLAMF6); etc. Antibodies capable of binding to each of such exemplary non-CD16 cell surface molecule are commercially available from Abcam plc and other sources, and may be readily adapted to the purposes of the present invention. In one embodiment, the molecules of the present invention that are capable of mediating the redirected killing of a target cell will bind an effector cell by immunospecifically binding an epitope of CD2 present on the surface of such effector cell. Molecules that specifically bind CD2 include the anti-CD2 antibody “CD2 mAb Lo-CD2a.” The amino acid sequence of the VH Domain of CD2 mAb Lo-CD2a (ATCC Accession No: 11423); SEQ ID NO:90) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of CD2 mAb Lo-CD2a (ATCC Accession No: 11423; SEQ ID NO:91) is shown below (CDRLresidues are shown underlined): In one embodiment, the molecules of the present invention that are capable of mediating the redirected killing of a target cell will bind an effector cell by immunospecifically binding an epitope of CD8 present on the surface of such effector cell. Antibodies that specifically bind CD8 include the anti-CD8 antibodies “OKT8” and “TRX2.” The amino acid sequence of the VH Domain of OKT8 (SEQ ID NO:92) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of OKT8 (SEQ ID NO:93) is shown below (CDRLresidues are shown underlined): The amino acid sequence of the VH Domain of TRX2 (SEQ ID NO:94) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of TRX2 (SEQ ID NO:95) is shown below (CDRLresidues are shown underlined): The Disease Antigens of the present invention comprise cell surface antigens that are characteristic of a cancer cell (“Cancer Antigens”) as well as cell surface antigens that are characteristic of a pathogen cell or a cell infected by a pathogen (Pathogen-Associated Antigens”). As used herein, the term “Cancer Antigen” denotes an antigen that is characteristically expressed on the surface of a cancer cell, and that may thus be treated with an Antibody-Based Molecule or an Immunomodulatory Molecule. Examples of Cancer Antigens include, but are not limited to: 19.9 as found in colon cancer, gastric cancer mucins; 4.2; A33 (a colorectal carcinoma antigen; Almqvist, Y. (2006) “ Exemplary antibodies, whose VH and VL Domains may be used to construct the binding molecules of the present invention that are capable of binding a Cancer Antigen arrayed on the surface of a cancer cell and mediating the redirected killing of such cancer cells are listed in Table 11, additional antibodies that may be used to construct molecules capable of binding a Cancer Antigen arrayed on the surface of a cancer cell and mediating the redirected killing of such cancer cells are provided below. B7-H3 is a Cancer Antigen that is over-expressed on a wide variety of solid tumor types and is a member of the B7 family of molecules that are involved in immune regulation (see, U.S. Pat. No. 8,802,091; US 2014/0328750; US 2013/0149236; Loo, D. et al. (2012) “ B7-H3 has also been found to co-stimulate CD4+ and CD8+ T-cell proliferation. B7-H3 also stimulates IFN-γ production and CD8+ lytic activity (Chapoval, A. et al. (2001) “ Preferred B7-H3-binding molecules possess the VL and/or VH Domains, of humanized anti-human B7-H3 monoclonal antibody “B7-H3 mAb-B,” “B7-H3 mAb-C,” “B7-H3 mAb-D,” and more preferably possess 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of such anti-B7-H3 monoclonal antibodies. Upon humanization, antibody B7-H3 mAb-B yielded two variant VH Domains, B7-H3 mAb-B VIII and B7-H3 mAb-B VH2; and two variant VL Domains B7-113 mAb-B VL1 and B7-H3 mAb-B VL2, which may be used in any combination of VH/VL Domains to yield a functional B7-H3 Binding Domain. The amino acid sequence of the VH Domain of B7-H3 mAb-B VIII (SEQ ID NO:96) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VH Domain of B7-H3 mAb-B VH2 (SEQ ID NO:97) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of B7-H3 mAb-B VL1 (SEQ ID NO:98) is shown below (CDRLresidues are shown underlined). The amino acid sequence of the VL Domain of B7-H3 mAb-B VL2 (SEQ ID NO:99) is shown below (CDRLresidues are shown underlined). The amino acid sequence of the VH Domain of humanized B7-H3 mAb-C (SEQ ID NO:100) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of humanized B7-H3 mAb-C (SEQ ID NO:101) is shown below (CDRLresidues are shown underlined). The amino acid sequence of the VH Domain of B7-H3 mAb-D (SEQ ID NO:102) is shown below (CDRHresidues are shown underlined). The amino acid sequence of the VL Domain of B7-H3 mAb-D (SEQ ID NO:103) is shown below (CDRLresidues are shown underlined). Particularly preferred, are B7-H3-binding molecules which possess a humanized VH and/or VL Domain including but not limited to “Enoblituzumab” (also known as MGA271; CAS Reg No. 1353485-38-7). Enoblituzumab is an Fc-optimized monoclonal antibody that binds to HER2/neu and mediates enhanced ADCC activity. The amino acid sequences of the complete Heavy and Light Chains of Enoblituzumab are known in the art (see., e.g., WHO Drug Information, 2017, Recommended INN: List 77, 31(1):49). The amino acid sequence of the VH Domain of Enoblituzumab (SEQ ID NO:104) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of Enoblituzumab (SEQ ID NO:105) is shown below (CDRLresidues are shown underlined): In addition to the above-identified preferred anti-B7-H3 Binding Molecules, the invention contemplates the use of any of the following anti-B7-H3 Binding Molecules: LUCA1; BLA8; PA20; or SKN2 (see, U.S. Pat. Nos. 7,527,969; 8,779,098 and PCT Patent Publication WO 2004/001381); M30; cM30; M30-H1-L1; M30-H1-L2; M30-H1-L3; M30-H1-L4; M30-H1-L5; M30-H1-L6; M30-H1-L7; M30-H4-L1; M30-H4-L2; M30-H4-L3; and M30-H4-L4 (see, US Patent Publication 2013/0078234 and PCT Patent Publication WO 2012/147713); and 8119 (see U.S. Pat. Nos. 7,666,424; 7,737,258; 7,740,845; 8,148,154; 8,414,892; 8,501,471; 9,062,110; US Patent Publication 2010/0143245 and PCT Patent Publication WO 2008/116219). The present invention specifically includes and encompasses CD16 x B7-H3 Binding Molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of any of B7-H3 mAb-B, B7-H3 mAb-B VH1, B7-H3 mAb-B VH2, B7-H3 mAb-B VL1, B7-H3 mAb-B VL2, B7-H3 mAb-C, B7-H3 mAb-D, or Enoblituzumab, or any of the other anti-B7-H3 antibodies provided herein; and more preferably possess 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of such anti-B7-H3 monoclonal antibodies. Carcinoembryonic Antigen-Related Cell Adhesion Molecules 5 (CEACAM5) and 6 (CEACAM6) have been found to be associated with various types of cancers including medullary thyroid cancer, colorectal cancer, pancreatic cancer, hepatocellular carcinoma, gastric cancer, lung cancer, head and neck cancers, urinary bladder cancer, prostate cancer, uterine cancer, endometrial cancer, breast cancer, hematopoietic cancer, leukemia and ovarian cancer (PCT Pubmication No. WO 2011/034660), and particularly colorectal, gastrointestinal, pancreatic, non-small cell lung cancer (NSCL), breast, thyroid, stomach, ovarian and uterine carcinomas (Zheng, C. et al. (2011) “ CEACAM5 has been found to be overexpressed in 90% of gastrointestinal, colorectal and pancreatic cancers, 70% of non-small cell lung cancer cells and 50% of breast cancers (Thompson, J. A. et al. (1991) “ The amino acid sequence of the VH Domain of the humanized anti-CEACAM5/ANTI-CEACAM6 antibody 16C3 (EP 2585476) (SEQ ID NO:106) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of the humanized anti-CEACAM5/ANTI-CEACAM6 antibody 16C3 (EP 2585476) (SEQ ID NO:107) is shown below (CDRLresidues are shown underlined): The amino acid sequence of the VH Domain of the humanized anti-CEACAM5/CEACAM6 antibody hMN15 (WO 2011/034660) (SEQ ID NO:108) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of the humanized anti-CEACAM5/CEACAM6 antibody hMN15 (WO 2011/034660) (SEQ ID NO:109) is shown below (CDRLresidues are shown underlined): The present invention specifically includes and encompasses CD16 x CEACAM5/CEACAM6 Binding Molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of the anti-CEACAM5/CEACAM6 monoclonal antibodies 16C3 or hMN15. Epidermal Growth Factor Receptor (EGFR) is a Cancer Antigen of certain metastatic colorectal cancer, metastatic non-small cell lung cancer and head and neck cancer. Exemplary antibodies that bind human EGRF are “Cetuximab” and “Panitumumab.” Cetuximab is a recombinant human-mouse chimeric epidermal growth factor receptor (EGFR) IgG1 monoclonal antibody (Govindan R. (2004) “ The amino acid sequence of the VH Domain of the chimeric anti-EGFR antibody Cetuximab (SEQ ID NO:110) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of the chimeric anti-EGFR antibody Cetuximab (SEQ ID NO:111) is shown below (CDRLresidues are shown underlined): The amino acid sequence of the VH Domain of Panitumumab (SEQ ID NO:112) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of Panitumumab (SEQ ID NO:113) is shown below (CDRLresidues are shown underlined): The present invention specifically includes and encompasses CD16 x EGFR Binding Molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of the anti-EGFR monoclonal antibodies Cetuximab or Panitumumab. The receptor tyrosine kinase, Ephrin type-A receptor 2 (EphA2) is normally expressed at sites of cell-to-cell contact in adult epithelial tissues, however, recent studies have shown that it is also overexpressed in various types of epithelial carcinomas, with the greatest level of EphA2 expression observed in metastatic lesions. High expression levels of EphA2 have been found in a wide range of cancers and in numerous cancer cell lines, including prostate cancer, breast cancer, non-small cell lung cancer and melanoma (Xu, J. et al. (2014) “ The amino acid sequence of the VH Domain of EphA2 mAb 1 (SEQ ID NO:114) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of EphA2 mAb 1 (SEQ ID NO:115) is shown below (CDRLresidues are shown underlined): The amino acid sequence of the VH Domain of EphA2 mAb 2 (SEQ ID NO:116) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of EphA2 mAb 2 (SEQ ID NO:117) is shown below (CDRLresidues are shown underlined): The amino acid sequence of the VH Domain of EphA2 mAb 3 (SEQ ID NO:118) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of EphA2 mAb 3 (SEQ ID NO:119) is shown below (CDRLresidues are shown underlined): The present invention specifically includes and encompasses CD16 x EphA2 Binding Molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRL5 of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of anti-EphA2 monoclonal antibodies EphA2 mAb 1, EphA2 mAb 2 and EphA2 mAb 3. The 43 kD transmembrane glycoprotein A33 (gpA33) is expressed in >95% of all colorectal carcinomas (Heath, J. K. et al. (1997) “ The amino acid sequence of the VH Domain of gpA33 mAb 1 (SEQ ID NO:120) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of gpA33 mAb 1 (SEQ ID NO:121) is shown below (CDRLresidues are shown underlined): The present invention specifically includes and encompasses CD16 x gpA33 Binding Molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of anti-gpA33 monoclonal antibodies gpA33 mAb 1, or of any of the anti-gpA33 monoclonal antibodies provided in WO 2015/026894. HER2/neu is a 185 kDa receptor protein that was originally identified as the product of the transforming gene from neuroblastomas of chemically treated rats. HER2/neu has been extensively investigated because of its role in several human carcinomas and in mammalian development (Hynes et al. (1994) Biochim. Biophys. Acta 1198:165-184; Dougall et al. (1994) Oncogene 9:2109-2123; Lee et al. (1995) Nature 378:394-398). Exemplary antibodies that bind human HER2/neu include “Margetuximab,” “Trastuzumab” and “Pertuzumab.” Margetuximab (also known as MGAH22; CAS Reg No. 1350624-75-7) is an Fc-optimized monoclonal antibody that binds to HER2/neu and mediates enhanced ADCC activity. Trastuzumab (also known as rhuMAB4D5, and marketed as H The present invention specifically includes and encompasses CD16 x HER2/neu Binding Molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of the anti-HER2/neu monoclonal antibodies Margetuximab, Trastuzumab or Pertuzumab. The amino acid sequence of the VH Domain of Margetuximab (SEQ ID NO:122 is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of Margetuximab (SEQ ID NO:123) is shown below (CDRLresidues are shown underlined): The amino acid sequences of the complete Heavy and Light Chains of Margetuximab are known in the art (see., e.g., WHO Drug Information, 2014, Recommended INN: List 71, 28(1):93-94). The amino acid sequence of the VH Domain of Trastuzumab (SEQ ID NO:124) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of Trastuzumab (SEQ ID NO:125) is shown below (CDRLresidues are shown underlined): The amino acid sequence of the VH Domain of Pertuzumab (SEQ ID NO:126) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of Pertuzumab (SEQ ID NO:127) is shown below (CDRLresidues are shown underlined): In addition to the above-identified preferred anti-HER2/neu Binding Molecules, the invention includes and encompasses CD16 x HER2/neu Binding Molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of any of the following anti-HER2/neu Binding Molecules: 1.44.1; 1.140; 1.43; 1.14.1; 1.100.1; 1.96; 1.18.1; 1.20; 1.39; 1.24; and 1.71.3 (U.S. Pat. Nos. 8,350,011; 8,858,942; and PCT Patent Publication WO 2008/019290); F5 and C1 (U.S. Pat. Nos. 7,892,554; 8,173,424; 8,974,792; and PCT Patent Publication WO 99/55367); and also the anti-HER2/neu Binding Molecules of US Patent Publication US2013017114 and PCT Patent Publications WO2011/147986 and WO 2012/143524). The present invention specifically includes and encompasses CD16 x HER2/neu Binding Molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of any of Margetuximab, Trastuzumab or Pertuzumab, or any of the other anti-HER2/neu antibodies provided herein; and more preferably possess 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of such anti-HER2/neu monoclonal antibodies. VEGF-A is a chemical signal that stimulates angiogenesis in a variety of diseases, especially in certain metastatic cancers such as metastatic colon cancer, and in certain lung cancers, renal cancers, ovarian cancers, and glioblastoma multiforme of the brain. An exemplary antibody that binds to human VEGF-A is “Bevacizumab” (Avastin®). Bevacizumab is a recombinant humanized IgG1 monoclonal antibody (Midgley, R. et al. (2005) “ The amino acid sequence of the VH Domain of Bevacizumab (SEQ ID NO:128) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of Bevacizumab (SEQ ID NO:129) is shown below (CDRLresidues are shown underlined): The present invention specifically includes and encompasses CD16 x VEGF Binding Molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRL5 of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of the anti-VEGF monoclonal antibody Bevacizumab. The oncofetal protein, 5T4, is a tumor-associated protein displayed on the cell membrane of many carcinomas, including kidney, colon, prostate, lung, carcinoma and in acute lymphoblastic leukemia (see, Boghaert, E. R. et al. (2008) “ The amino acid sequence of the VH Domain of 5T4 mAb 1 (SEQ ID NO:130) is shown below (CDR residues are shown underlined): The amino acid sequence of the VL Domain of an 5T4 mAb 1 (SEQ ID NO:131) is shown below (CDR residues are shown underlined): The amino acid sequence of the VH Domain of 5T4 mAb 2 (SEQ ID NO:132) is shown below (CDR residues are shown underlined): The amino acid sequence of the VL Domain of 5T4 mAb 2 (SEQ ID NO:133) is shown below (CDR residues are shown underlined): The present invention specifically includes and encompasses CD16 x 5T4 Binding Molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of the anti-5T4 monoclonal antibodies 5T4 mAb 1 or 5T4 mAb 2, or of any of the anti-5T4 antibodies provided in WO 2013/041687 or WO 2015/184203. The present application additionally specifically includes and encompasses CD16 x 5T4 Trispecific Binding Molecules that are capable of binding to 5T4, to CD16 and to CD8, and particularly such trispecific binding molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of the anti-5T4 monoclonal antibodies 5T4 mAb 1 or 5T4 mAb 2 or of any of the anti-5T4 monoclonal antibodies provided in WO 2015/184203, and/or the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of any of the anti-CD8 monoclonal antibodies provided herein. Interleukin-13 Receptor α2 (IL13Rα2) is overexpressed in a variety of cancers, including glioblastoma, colorectal cancer, cervical cancer, pancreatic cencer, multiple melanoma, osteosarcoma, leukemia, lymphoma, prostate cancer and lung cancer (PCT Pubmication No. WO 2008/146911; Brown, C. E. et al. (2013) “ The amino acid sequence of the VH Domain of hu08 (SEQ ID NO:134) is shown below (CDR residues are shown underlined): The amino acid sequence of the VL Domain of hu08 (SEQ ID NO:135) is shown below (CDR residues are shown underlined): The present invention specifically includes and encompasses CD16 x IL13Rα2 Binding Molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of the anti-IL13Rα2 monoclonal antibody hu08. CD123 (interleukin 3 receptor alpha, IL-3Ra) is a 40 kDa molecule and is part of the interleukin 3 receptor complex (Stomski, F. C. et al. (1996) “ An exemplary antibody that binds to human CD123, and that may be employed in the present invention, is “CD123 mAb 1” (see, e.g., PCT Patent Publication WO 2015/026892). The amino acid sequence of the VH Domain of CD123 mAb 1 (SEQ ID NO:136) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of CD123 mAb 1 (SEQ ID NO:137) is shown below (CDRLresidues are shown underlined): The present invention specifically includes and encompasses CD16 x CD123 Binding Molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of the anti-CD123 monoclonal antibody CD123 mAb 1 or any of the anti-CD123 antibodies disclosed in US 2017/081424 and WO 2016/036937. CD19 (B lymphocyte surface antigen B4, Genbank accession number M28170) is a component of the B cell-receptor (BCR) complex, and is a positive regulator of B cell signaling that modulates the threshold for B cell activation and humoral immunity. CD19 is one of the most ubiquitously expressed antigens in the B cell lineage and is expressed on >95% of B cell malignancies, including acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL), and non-Hodgkin's Lymphoma (NHL). Notably, CD19 expression is maintained on B cell lymphomas that become resistant to anti-CD20 therapy (Davis et al. (1999) “ An exemplary antibody that binds to human CD19, and that may be employed in the present invention, is the anti-CD19 antibody disclosed in WO 2016/048938 (referred to herein as “CD19 mAb 1”). The amino acid sequence of the VH Domain of CD19 mAb 1 (SEQ ID NO:123) is shown below (CDRHresidues are shown underlined): The amino acid sequence of the VL Domain of CD19 mAb 1 (SEQ ID NO:139) is shown below (CDRLresidues are shown underlined): The present invention specifically includes and encompasses CD16 x CD19 Binding Molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRL5 of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of the anti-CD19 monoclonal antibody CD19 mAb 1, or any of the anti-CD19 antibodies disclosed in U.S. Pat. No. 7,112,324. As used herein, the term “Pathogen-Associated Antigen” denotes an antigen that is characteristically expressed on the surface of a pathogen-infected cell, and that may thus be treated with an Antibody-Based Molecule or an Immunomodulatory Molecule. Examples of Pathogen-Associated Antigens include, but are not limited to antigens expressed on the surface of a cell infected with: a Herpes Simplex Virus (e.g., infected cell protein (ICP)47, gD, etc.), a varicella-zoster virus, a Kaposi's sarcoma-associated herpesvirus, an Epstein-Barr Virus (e.g., LMP-1, LMP-2A, LMP-2B, etc.), a Cytomegalovirus (e.g., UL11, etc.), Human Immunodeficiency Virus (e.g., env proteins gp160, gp120, gp41, etc.), a Human Papillomavirus (e.g., E6, E7, etc.), a human T-cell leukemia virus (e.g., env proteins gp64, gp46, gp21, etc.), Hepatitis A Virus, Hepatitis B Virus, Hepatitis C Virus, Vesicular Stomatitis Virus (VSV), Bacilli, Exemplary antibodies, whose VH and VL Domains may be used to construct molecules capable of binding a Pathogen-Associated Antigen arrayed on the surface of a pathogen-infected cell are antibodies are provided below, additional antibodies are known in the art. The env protein of HIV is an exemplary Pathogen-Associated Antigen, and antibodies that bind the env protein of HIV are exemplary of antibodies capable of binding a Pathogen-Associated Antigen. The initial step in HIV-1 infection occurs with the binding of cell surface CD4 to trimeric HIV-1 envelope glycoproteins (env), a heterodimer of a transmembrane glycoprotein (gp41) and a surface glycoprotein (gp120). The gp120 and gp41 glycoproteins are initially synthesized as a single gp160 polypeptide that is subsequently cleaved to generate the non-covalently associated gp120/gp41 complex. The ectodomain of env is a heterodimer with mass of approximately 140 kDa, composed of the entire gp120 component, and approximately 20 kDa of gp41 (Harris, A. et al. (2011) “ Antibody 7B2 (Genbank accession numbers JX188438 and JX188439) is an anti-HIV env human IgG1 antibody that binds HIV gp41 at 598-604 in the immunodominant helix-loop-helix region of the molecule (Sadraeian, M. et al. (2017) “ The amino acid sequence of the VH Domain of 7B2 (SEQ ID NO:140) is shown below (CDR residues are shown underlined): The amino acid sequence of the VL Domain of 7B2 (SEQ ID NO:141) is shown below (CDR residues are shown underlined): Monoclonal antibody A32 recognizes a conformational epitope in the C1 region of HIV-1 Env gp120 (Wyatt et al. (1995) “ Multiple VH Domains of Antibody A32 have been reported in the art that possess minor changes in framework regions 1 and/or 4 reported (see, e.g., Protein Data Base Accession number PDB: 4YBL_H, US 2015/0239961 and WO 2006/044410). Any of these variant Antibody A32 VH Domains may be employed in accordance with the present invention. The amino acid sequence of an illustrative VH Domain of A32 (SEQ ID NO:142) is shown below (CDR residues are shown underlined): The amino acid sequence of the VL Domain of A32 (SEQ ID NO:143) is shown below (CDR residues are shown underlined): The amino acid sequence of the VL Domain of A32 (SEQ ID NO:143) may be employed with the illustrative VH Domain of A32 (SEQ ID NO:142) or with any of the variant Antibody A32 VH Domains (see, e.g., Protein Data Base Accession number PDB: 4YBL_H, US 2015/0239961 and WO 2006/044410) to form an anti-HIV-1 Env gp120 Epitope Binding Site. The present invention specifically includes and encompasses CD16 x HIV Binding Molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of the anti-HIV monoclonal antibodies 7B2 or A32, or of any of the anti-HIV antibodies disclosed in WO 2016196975, WO 2016/149710, WO 2016/149698, WO 2016/149695, WO 2015/048610, WO 2012/030904, WO 2013/163427, WO 2013/192589, WO 2014/063059, WO 20170/11413, WO 2016/054101, WO 2014/159940, or WO 2017/011414. The present application additionally specifically includes and encompasses CD16 x HIV Binding Molecules that are capable of binding to HIV, to CD16 and to CD8, and particularly such trispecific binding molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of the anti-HIV monoclonal antibodies 7B2 or A32 or of any of the anti-HIV monoclonal antibodies provided in WO 2015/184203, WO 2016/054101, WO 2017/011413, WO 2017/011414. A further illustrative Pathogen-Associated Antigen is RSV glycoprotein F. An exemplary anti-RSV glycoprotein F antibody is Palivizumab (see, e.g., Protein Data Bank (PDB) ID No. 2HWZ). Alternative anti-RSV glycoprotein F antibodies include motavizumab (see, e.g., PDB ID No. 3IXT) and a variant of palivizumab (also referred to herein as “vPalivizumab”) that has been engineered to remove a cysteine residue from palivizumab' s CDRL1. The amino acid sequence of the VH Domain of the variant of palivizumab (SEQ ID NO:144) is shown below (CDR residues are shown underlined): The amino acid sequence of the VL Domain of the variant of palivizumab (SEQ ID NO:145) is shown below (CDR residues are shown underlined): The present invention specifically includes and encompasses CD16 x RSV Binding Molecules that comprise the VL and/or VH Domain, and/or 1, 2 or all 3 of the CDRLs of the VL Region and/or 1, 2 or all 3 of the CDRHs of the VH Domain of the anti-RSV monoclonal antibody palivizumab or vPalivizumab. The principles of the present invention are illustrated by a series of exemplary CD16 x DA Binding Molecules incorporating different murine or humanized anti-CD16 binding domains and having a Binding Domain that is immunospecific for a Disease Antigen. The covalent diabody structures and sequences of such illustrative CD16 x DA Binding Molecules are summarized in Table 12, and are described in detail below. As will be recognized, analogous diabodies and other bispecific molecules may likewise be constructed (by employing the VL and VH domains of desired antibodies in lieu of the VL and VH domains used in the illustrative CD16 x DA Binding Molecules. The CD16 x HER2/neu Binding Molecule designated “DART-A” is a first illustrative CD16 x DA Binding Molecule. DART-A is an Fc Domain-containing, bispecific diabody capable of binding CD16 and the HER2/neu cancer antigen. DART-A is composed of three polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of anti-human CD16 antibody CD16-M1 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of Trastuzumab (and is thus immunospecific for an epitope of the HER2/neu Cancer Antigen). The three polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of the HER2/neu Cancer Antigen (see, e.g., The first polypeptide chain of DART-A has the amino acid sequence of SEQ ID NO:151: Residues 1-107 of the first polypeptide chain (SEQ ID NO:151) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of Trastuzumab (SEQ ID NO:125). Residues 108-115 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-233 of the first polypeptide chain correspond to the VH Domain of anti-human CD16 antibody CD16-M1 (SEQ ID NO:64). Residues 234-238 correspond to a linker (SEQ ID NO:21, underlined). Residues 239-266 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). Residues 267-279 of the first polypeptide chain correspond to a linker (SEQ ID NO:43). Residues 280-496 of the first polypeptide chain correspond to a “knob-bearing” (SEQ ID NO:51), in which the final residue is lysine. The second polypeptide chain of DART-A has the amino acid sequence of SEQ ID NO:152: Residues 1-107 of the second polypeptide chain (SEQ ID NO:152) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of anti-human CD16 antibody CD16-M1 (SEQ ID NO:65). Residues 108-115 (double underlined) of the second polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-235 of the second polypeptide chain correspond to the VH Domain of Trastuzumab (SEQ ID NO:124). Residues 236-240 of the second polypeptide chain correspond to a linker (SEQ ID NO:21, underlined). Residues 241-268 of the second polypeptide chain correspond to a cysteine-containing K-coil (SEQ ID NO:32). The third polypeptide chain of the DART-A has the amino acid sequence of SEQ ID NO:153: Residues 1-10 of the third polypeptide chain (SEQ ID NO:153) of such illustrative CD16 x DA Binding Molecule correspond to a linker (SEQ ID NO:40). Residues 11-227 of the third polypeptide chain correspond to a “hole-bearing” IgG1 CH2-CH3 Domain (SEQ ID NO:53), containing the H435R substitution (shown underlined), and in which the final residue is lysine. As stated above, the H435R substitution eliminates the ability of the molecule to bind to bind protein A. As will be recognized, the third polypeptide chain of DART-A does not contain any Epitope Binding sites and may thus be employed in various CD16 x DA Binding Molecules. Accordingly, the third polypeptide chain of DART-A is referred to as a “Common Diabody Polypeptide Chain.” The CD16 x Her2/neu Binding Molecule designated “DART-B” is a further illustrative CD16 x DA Binding Molecule. DART-B is an Fc Domain-containing, bispecific diabody capable of binding CD16 and the HER2/neu Cancer Antigen. DART-B is composed of three polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of anti-human CD16 antibody CD16-M2 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of Trastuzumab (and is thus immunospecific for an epitope of the HER2/neu Cancer Antigen). The three polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of the HER2/neu Cancer Antigen (see, e.g., The first polypeptide chain of DART-B has the amino acid sequence of SEQ ID NO:154: Residues 1-107 of the first polypeptide chain (SEQ ID NO:154) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of Trastuzumab (SEQ ID NO:125). Residues 108-115 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-234 of the first polypeptide chain correspond to the VH Domain of anti-human CD16 antibody CD16-M2 (SEQ ID NO:75). Residues 235-239 correspond to a linker (SEQ ID NO:21, underlined). Residues 240-267 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). Residues 268-280 of the first polypeptide chain correspond to a linker (SEQ ID NO:43). Residues 281-497 of the first polypeptide chain correspond to a “knob-bearing” IgG1 CH2-CH3 Domain (SEQ ID NO:51), in which the final residue is lysine. The second polypeptide chain of DART-B has the amino acid sequence of SEQ ID NO:155: Residues 1-107 of the second polypeptide chain (SEQ ID NO:155) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of anti-human CD16 antibody CD16-M2 (SEQ ID NO:76). Residues 108-115 (double underlined) of the second polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-235 of the second polypeptide chain correspond to the VH Domain of Trastuzumab (SEQ ID NO:124). Residues 236-240 of the second polypeptide chain correspond to a linker (SEQ ID NO: 21, underlined). Residues 241-268 of the second polypeptide chain correspond to a cysteine-containing K-coil (SEQ ID NO:32). The third polypeptide chain of DART-B has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The CD16 x HER2/neu Binding Molecule designated “DART-C” is a further illustrative CD16 x DA Binding Molecule. DART-C is an Fc Domain-containing, bispecific diabody capable of binding CD16 and the HER2/neu Cancer Antigen. DART-C is composed of three polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of humanized anti-human CD16 antibody hCD16-M1 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of Trastuzumab (and is thus immunospecific for an epitope of the HER2/neu Cancer Antigen). The three polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of the HER2/neu Cancer Antigen (see, e.g., The first polypeptide chain of DART-C has the amino acid sequence of SEQ ID NO:156: Residues 1-107 of the first polypeptide chain (SEQ ID NO:156) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of Trastuzumab (SEQ ID NO:125). Residues 108-115 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-233 of the first polypeptide chain correspond to the VH Domain of the humanized anti-human CD16 antibody hCD16-M1 (SEQ ID NO:72). Residues 234-238 correspond to a linker (SEQ ID NO:21, underlined). Residues 239-266 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). Residues 267-279 of the first polypeptide chain correspond to a linker (SEQ ID NO:43). Residues 280-496 of the first polypeptide chain correspond to a “knob-bearing” IgG1 CH2-CH3 Domain (SEQ ID NO:51), in which the final residue is lysine. The second polypeptide chain of DART-C has the amino acid sequence of SEQ ID NO:157: Residues 1-107 of the second polypeptide chain (SEQ ID NO:157) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of humanized anti-human CD16 antibody hCD16-M1 (SEQ ID NO:73). Residues 108-115 (double underlined) of the second polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-235 of the second polypeptide chain correspond to the VH Domain of Trastuzumab (SEQ ID NO:124). Residues 236-240 of the second polypeptide chain correspond to a linker (SEQ ID NO:21, underlined). Residues 241-268 of the second polypeptide chain correspond to a cysteine-containing K-coil (SEQ ID NO:32). The third polypeptide chain of DART-C has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The CD16 x HER2/neu Binding Molecule designated “DART-D” is a further illustrative CD16 x DA Binding Molecule. DART-D is an Fc Domain-containing, bispecific diabody capable of binding CD16 and the HER2/neu Cancer Antigen. DART-D is composed of three polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of humanized anti-human CD16 antibody hCD16-M2 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of Trastuzumab (and is thus immunospecific for an epitope of the HER2/neu Cancer Antigen). The three polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of the HER2/neu Cancer Antigen (see, e.g., The first polypeptide chain of DART-D has the amino acid sequence of SEQ ID NO:158: Residues 1-107 of the first polypeptide chain (SEQ ID NO:158) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of Trastuzumab (SEQ ID NO:125). Residues 108-115 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-234 of the first polypeptide chain correspond to the VH1 Domain of the humanized anti-human CD16 antibody hCD16-M2 (SEQ ID NO:83). Residues 235-239 correspond to a linker (SEQ ID NO:21, underlined). Residues 240-267 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). Residues 268-280 of the first polypeptide chain correspond to a linker (SEQ ID NO:43). Residues 281-497 of the first polypeptide chain correspond to a “knob-bearing” IgG1 CH2-CH3 Domain (SEQ ID NO:51), in which the final residue is lysine. The second polypeptide chain of DART-D has the amino acid sequence of SEQ ID NO:159: Residues 1-107 of the second polypeptide chain (SEQ ID NO:159) of such illustrative CD16 x DA Binding Molecule correspond to the VL1 Domain of humanized anti-human CD16 antibody hCD16-M2 (SEQ ID NO:85). Residues 108-115 (double underlined) of the second polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-235 of the second polypeptide chain correspond to the VH Domain of Trastuzumab (SEQ ID NO:124). Residues 236-240 of the second polypeptide chain correspond to a linker (SEQ ID NO:21, underlined). Residues 241-268 of the second polypeptide chain correspond to a cysteine-containing K-coil (SEQ ID NO:32). The third polypeptide chain of DART-D has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The CD16 x HER2/neu Binding Molecule designated “DART-E” is a further illustrative CD16 x DA Binding Molecule. DART-E is a bispecific diabody capable of binding CD16 and the HER2/neu Cancer Antigen. DART-E is composed of three polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of humanized anti-human CD16 antibody hCD16-M2 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of Trastuzumab (and is thus immunospecific for an epitope of the HER2/neu Cancer Antigen). The three polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of the HER2/neu Cancer Antigen (see, e.g., The first polypeptide chain of DART-E has the amino acid sequence of SEQ ID NO:160: Residues 1-107 of the first polypeptide chain (SEQ ID NO:160) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of Trastuzumab (SEQ ID NO:125). Residues 108-115 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-234 of the first polypeptide chain correspond to the VH2 Domain of the humanized anti-human CD16 antibody hCD16-M2 (SEQ ID NO:84). Residues 235-239 correspond to a linker (SEQ ID NO:21, underlined). Residues 240-267 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). Residues 268-280 of the first polypeptide chain correspond to a linker (SEQ ID NO:43). Residues 281-497 of the first polypeptide chain correspond to a “knob-bearing” IgG1 CH2-CH3 Domain (SEQ ID NO:51), in which the final residue is lysine. The second polypeptide chain of DART-E is identical in sequence to the second polypeptide chain of DART-D (SEQ ID NO:159). The third polypeptide chain of DART-E has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The CD16 x HIV env Binding Molecule designated “DART-F” is a further illustrative CD16 x DA Binding Molecule. DART-F is a bispecific diabody capable of binding CD16 and the HIV env protein. DART-F is composed of three polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of humanized anti-human CD16 antibody hCD16-M1 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of Antibody A32 (and is thus immunospecific for an epitope of the HIV env protein). The three polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of the HIV env protein (see, e.g., The first polypeptide chain of DART-F has the amino acid sequence of SEQ ID NO:161: Residues 1-110 of the first polypeptide chain (SEQ ID NO:161) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of Antibody A32 (SEQ ID NO:143). Residues 111-118 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 119-236 of the first polypeptide chain correspond to the VH Domain of the humanized anti-human CD16 antibody hCD16-M1 (SEQ ID NO:72). Residues 237-241 correspond to a linker (SEQ ID NO:21, underlined). Residues 242-269 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). Residues 270-282 of the first polypeptide chain correspond to a linker (SEQ ID NO:43). Residues 283-499 of the first polypeptide chain correspond to a “knob-bearing” IgG1 CH2-CH3 Domain (SEQ ID NO:51), in which the final residue is lysine. The second polypeptide chain of DART-F has the amino acid sequence of SEQ ID NO:162: Residues 1-107 of the second polypeptide chain (SEQ ID NO:162) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of humanized anti-human CD16 antibody hCD16-M1 (SEQ ID NO:73). Residues 108-115 (double underlined) of the second polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-238 of the second polypeptide chain correspond to the VH Domain of Antibody A32 (SEQ ID NO:142). Residues 239-243 of the second polypeptide chain correspond to a linker (SEQ ID NO:21, underlined). Residues 244-271 of the second polypeptide chain correspond to a cysteine-containing K-coil (SEQ ID NO:32). The third polypeptide chain of DART-F has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The CD16 x HIV env Binding Molecule designated “DART-G” is a further illustrative CD16 x DA Binding Molecule. DART-G is a bispecific diabody capable of binding CD16 and the HIV env protein. DART-G is composed of three polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of humanized anti-human CD16 antibody hCD16-M2 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of Antibody A32 (and is thus immunospecific for an epitope of the HIV env protein). The three polypeptide chains associate to form a covalently bonded DART® diabody composed capable of immunospecifically binding the epitope of CD16 and the epitope of the HIV env protein (see, e.g., The first polypeptide chain of the DART-G has the amino acid sequence of SEQ ID NO:163: Residues 1-110 of the first polypeptide chain (SEQ ID NO:163) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of Antibody A32 (SEQ ID NO:143). Residues 111-118 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 119-237 of the first polypeptide chain correspond to the VH Domain of the humanized anti-human CD16 antibody hCD16-M2 (SEQ ID NO:83). Residues 238-242 correspond to a linker (SEQ ID NO:21, underlined). Residues 243-270 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). Residues 271-283 of the first polypeptide chain correspond to a linker (SEQ ID NO:43). Residues 284-500 of the first polypeptide chain correspond to a “knob-bearing” IgG1 CH2-CH3 Domain (SEQ ID NO:51), in which the final residue is lysine. The second polypeptide chain of the DART-G has the amino acid sequence of SEQ ID NO:164: Residues 1-107 of the second polypeptide chain (SEQ ID NO:164) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of humanized anti-human CD16 antibody hCD16-M2 (SEQ ID NO:85). Residues 108-115 (double underlined) of the second polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-238 of the second polypeptide chain correspond to the VH Domain of Antibody A32 (SEQ ID NO:142). Residues 239-243 of the second polypeptide chain correspond to a linker (SEQ ID NO:21, underlined). Residues 244-271 of the second polypeptide chain correspond to a cysteine-containing K-coil (SEQ ID NO:32). The third polypeptide chain of DART-G has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The CD16 x HIV env Binding Molecule designated “DART-H” is a further illustrative CD16 x DA Binding Molecule. DART-H is a bispecific diabody capable of binding CD16 and the HIV env protein. DART-H is composed of three polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of humanized anti-human CD16 antibody hCD16-M1 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of Antibody 7B2 (and is thus immunospecific for an epitope of the HIV env protein). The three polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of the HIV env protein (see, e.g., The first polypeptide chain of DART-H has the amino acid sequence of SEQ ID NO:165: Residues 1-113 of the first polypeptide chain (SEQ ID NO:165) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of Antibody 7B2 (SEQ ID NO:141). Residues 114-121 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 122-239 of the first polypeptide chain correspond to the VH Domain of the humanized anti-human CD16 antibody hCD16-M1 (SEQ ID NO:72). Residues 240-244 correspond to a linker (SEQ ID NO:21, underlined). Residues 245-272 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). Residues 273-285 of the first polypeptide chain correspond to a linker (SEQ ID NO:43). Residues 286-502 of the first polypeptide chain correspond to a “knob-bearing” IgG1 CH2-CH3 Domain (SEQ ID NO:51), in which the final residue is lysine. The second polypeptide chain of DART-H has the amino acid sequence of SEQ ID NO:166: Residues 1-107 of the second polypeptide chain (SEQ ID NO:166) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of humanized anti-human CD16 antibody hCD16-M1 (SEQ ID NO:73). Residues 108-115 (double underlined) of the second polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-241 of the second polypeptide chain correspond to the VH Domain of Antibody 7B2 (SEQ ID NO:140). Residues 242-246 of the second polypeptide chain correspond to a linker (SEQ ID NO:21, underlined). Residues 247-274 of the second polypeptide chain correspond to a cysteine-containing K-coil (SEQ ID NO:32). The third polypeptide chain of DART-H has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The CD16 x HER2/neu Binding Molecule designated “DART-I” is a further illustrative CD16 x DA Binding Molecule. DART-I is similar to the above-described DART-C, but contains the VH of hCD16-M1A (comprising a mutated CDRH3). As indicated above, the VL Domain of hCD16-M1A has the same amino acid sequence as the VL Domain of hCD16-M1. The first polypeptide chain of DART-I has the amino acid sequence of SEQ ID NO:186: Residues 1-107 of the first polypeptide chain (SEQ ID NO:186) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of Trastuzumab (SEQ ID NO:125). Residues 108-115 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-233 of the first polypeptide chain correspond to the VH Domain of the optimized anti-human CD16 antibody hCD16-M1A (SEQ ID NO:58). Residues 234-238 correspond to a linker (SEQ ID NO:21, underlined). Residues 239-266 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). Residues 267-279 of the first polypeptide chain correspond to a linker (SEQ ID NO:43). Residues 280-496 of the first polypeptide chain correspond to a “knob-bearing” IgG1 CH2-CH3 Domain (SEQ ID NO:51), in which the final residue is lysine. Since the VL Domain of hCD16-M1A is the same as that of hCD16-M1, the amino acid sequence of the second polypeptide chain of DART-I is the same as that of the second polypeptide chain of the DART-C (i.e., SEQ ID NO:157). Similarly, the third polypeptide chain of DART-I has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The CD16 x HER2/neu Binding Molecule designated “DART-J” is a further illustrative CD16 x DA Binding Molecule. DART-J is similar to the above-described DART-C, but contains the VL of hCD16-M1B (comprising a mutated CDRL3). As indicated above, the VH Domain of hCD16-M1B has the same amino acid sequence as the VH Domain of hCD16-M1. Since the VH Domain of hCD16-M1B is the same as that of hCD16-M1, the amino acid sequence of the first polypeptide chain of DART-J is the same as that of the first polypeptide chain of the DART-C (i.e., SEQ ID NO:156). The second polypeptide chain of DART-J has the amino acid sequence of SEQ ID NO:187: Residues 1-107 of the second polypeptide chain (SEQ ID NO:157) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of humanized anti-human CD16 antibody hCD16-M1B (SEQ ID NO:59). Residues 108-115 (double underlined) of the second polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-235 of the second polypeptide chain correspond to the VH Domain of Trastuzumab (SEQ ID NO:124). Residues 236-240 of the second polypeptide chain correspond to a linker (SEQ ID NO:21, underlined). Residues 241-268 of the second polypeptide chain correspond to a cysteine-containing K-coil (SEQ ID NO:32). The third polypeptide chain of DART-J has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The CD16 x HER2/neu Binding Molecule designated “DART-K” is a further illustrative CD16 x DA Binding Molecule. DART-K is similar to the above-described DART-J, but also contains VH of hCD16-M1A (comprising a mutated CDRH3) Thus, DART-K contains the VL and VH of hCD16-M1AB (comprising a mutated CDRL3 and a mutated CDRH3). Since DART-K contains the VH of hCD16-M1A the amino acid sequence of the first polypeptide chain of DART-K is the same as that of the first polypeptide chain of the DART-I (i.e., SEQ ID NO:186). Since DART-K contains VL Domain of hCD16-M1B, the amino acid sequence of the second polypeptide chain of DART-K is the same as that of the second polypeptide chain of the DART-J (i.e., SEQ ID NO:187). The third polypeptide chain of DART-J has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The CD16 x CD19 Binding Molecule designated “DART-L” is another illustrative CD16 x DA Binding Molecule. DART-L is an Fc Domain-containing, bispecific diabody capable of binding CD16 and the CD19 B-cell tumor antigen. DART-L is composed of three polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of anti-human CD16 antibody hCD16-M1 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of CD19 mAb 1 (and is thus immunospecific for an epitope of the CD19 Cancer Antigen). The three polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of the CD19 Cancer Antigen (see, e.g., The first polypeptide chain of DART-L has the amino acid sequence of SEQ ID NO:188: Residues 1-106 of the first polypeptide chain (SEQ ID NO:188) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of CD19 mAb 1 (SEQ ID NO:139). Residues 107-114 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 115-232 of the first polypeptide chain correspond to the VH Domain of anti-human CD16 antibody hCD16-M1 (SEQ ID NO:72). Residues 233-238 correspond to a linker (SEQ ID NO:17, underlined). Residues 239-266 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). Residues 267-279 of the first polypeptide chain correspond to a linker (SEQ ID NO:43). Residues 280-496 of the first polypeptide chain correspond to a “knob-bearing” (SEQ ID NO:51), in which the final residue is lysine. The second polypeptide chain of DART-L has the amino acid sequence of SEQ ID NO:189: Residues 1-107 of the second polypeptide chain (SEQ ID NO:189) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of anti-human CD16 antibody hCD16-M1 (SEQ ID NO:73). Residues 108-115 (double underlined) of the second polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-235 of the second polypeptide chain correspond to the VH Domain of CD19 mAb 1 (SEQ ID NO:138). Residues 236-241 of the second polypeptide chain correspond to a linker (SEQ ID NO:17, underlined). Residues 242-269 of the second polypeptide chain correspond to a cysteine-containing K-coil (SEQ ID NO:32). The third polypeptide chain of DART-L has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The CD16 x CD19 Binding Molecule designated “DART-M” is a further illustrative CD16 x DA Binding Molecule. DART-M is similar to the above-described DART-L, but contains the VL of hCD16-M1B (comprising a mutated CDRL3). As indicated above, the VH Domain of hCD16-M1B has the same amino acid sequence as the VH Domain of hCD16-M1. Since the VH Domain of hCD16-M1B is the same as that of hCD16-M1, the amino acid sequence of the first polypeptide chain of DART-M is the same as that of the first polypeptide chain of the DART-L (i.e., SEQ ID NO:188). The second polypeptide chain of DART-M has the amino acid sequence of SEQ ID NO:190: Residues 1-107 of the second polypeptide chain (SEQ ID NO:190) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of anti-human CD16 antibody hCD16-M1B (SEQ ID NO:59). Residues 108-115 (double underlined) of the second polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-235 of the second polypeptide chain correspond to the VH Domain of CD19 mAb 1 (SEQ ID NO:138). Residues 236-241 of the second polypeptide chain correspond to a linker (SEQ ID NO:17, underlined). Residues 242-269 of the second polypeptide chain correspond to a cysteine-containing K-coil (SEQ ID NO:32). The third polypeptide chain of DART-M has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The CD16 x CD19 Binding Molecule designated “DART-N” is a further illustrative CD16 x DA Binding Molecule. DART-N is similar to the above-described DART-M, but also contains the VH of hCD16-M1A (comprising a mutated CDRH3). Thus, DART-N contains the VL and VH of hCD16-M1AB (comprising a mutated CDRL3 and a mutated CDRH3)). The first polypeptide chain of DART-N has the amino acid sequence of SEQ ID NO:191: Residues 1-106 of the first polypeptide chain (SEQ ID NO:191) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of CD19 mAb 1 (SEQ ID NO:139). Residues 107-114 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 115-232 of the first polypeptide chain correspond to the VH Domain of anti-human CD16 antibody hCD16-M1A (SEQ ID NO:58). Residues 233-238 correspond to a linker (SEQ ID NO:17, underlined). Residues 239-266 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). Residues 267-279 of the first polypeptide chain correspond to a linker (SEQ ID NO:43). Residues 280-496 of the first polypeptide chain correspond to a “knob-bearing” (SEQ ID NO:51), in which the final residue is lysine. Since DART-N contains VL Domain of hCD16-M1B, the amino acid sequence of the second polypeptide chain of DART-N is the same as that of the second polypeptide chain of the DART-M (i.e., SEQ ID NO:190). The third polypeptide chain of DART-N has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The CD16 x HER2/neu Binding Molecule designated “DART-1” is a further illustrative CD16 x DA Binding Molecule. DART-1 is a bispecific diabody capable of binding CD16 and the HER2/neu Cancer Antigen. DART-1 is essentially the same as DART-A, except that it possesses the VL and VH Domains of the humanized anti-human CD16 antibody h3G8 (see, e.g., U.S. Patent Publication No. 2004/0010124; PCT Publication No. WO 2017/142928; Li, W. et al. (2016) “ The first polypeptide chain of DART-1 has the amino acid sequence of SEQ ID NO:167: Residues 1-107 of the first polypeptide chain (SEQ ID NO:167) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of Trastuzumab (SEQ ID NO:125). Residues 108-115 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-233 of the first polypeptide chain correspond to the VH Domain of the humanized anti-human CD16 antibody h3G8 (SEQ ID NO:62). Residues 234-238 correspond to a linker (SEQ ID NO:21, underlined). Residues 239-266 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). Residues 267-279 of the first polypeptide chain correspond to a linker (SEQ ID NO:43). Residues 280-496 of the first polypeptide chain correspond to a “knob-bearing” IgG1 CH2-CH3 Domain (SEQ ID NO:51), in which the final residue is lysine. The second polypeptide chain of DART-1 has the amino acid sequence of SEQ ID NO:168: Residues 1-111 of the second polypeptide chain (SEQ ID NO:168) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of humanized anti-human CD16 antibody h3G8 (SEQ ID NO:63). Residues 112-119 (double underlined) of the second polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 120-239 of the second polypeptide chain correspond to the VH Domain of Trastuzumab (SEQ ID NO:124). Residues 240-244 of the second polypeptide chain correspond to a linker (SEQ ID NO:21, underlined). Residues 244-272 of the second polypeptide chain correspond to a cysteine-containing K-coil (SEQ ID NO:32). The third polypeptide chain of DART-1 has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The CD16 x RSV Binding Molecule designated “DART-2” is a further illustrative CD16 x DA Binding Molecule. DART-2 is a bispecific diabody capable of binding CD16 and the RSV glycoprotein F. DART-2 is essentially the same as the DART-1, except that it possesses the VH and VL Domains of the anti-RSV glycoprotein F antibody vPalivizumab (SEQ ID NO:144 and SEQ ID NO:145, respectively) instead of the VH and VL Domains of Trastuzumab (SEQ ID NO:124 and SEQ ID NO:125, respectively) that are present in DART-1 Diabody. Thus, DART-2 is composed of three polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of the humanized anti-human CD16 antibody h3G8 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of vPalivizumab (and is thus immunospecific for an epitope of the RSV glycoprotein F). The third polypeptide chain has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The three polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of RSV glycoprotein F (see, e.g., The CD16 x RSV Binding Molecule designated “DART-3” is a further illustrative CD16 x DA Binding Molecule. The CD16 x RSV DART-3 is a bispecific diabody capable of binding CD16 and the RSV glycoprotein F. DART-3 is composed of three polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of the humanized anti-human CD16 antibody hCD16-M1 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of vPalivizumab (and is thus immunospecific for an epitope of the RSV glycoprotein F). The three polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of the RSV glycoprotein F (see, e.g., The first polypeptide chain of DART-3 has the amino acid sequence of SEQ ID NO:169: Residues 1-106 of the first polypeptide chain (SEQ ID NO:169) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of vPalivizumab (SEQ ID NO:145). Residues 107-114 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 115-232 of the first polypeptide chain correspond to the VH Domain of the humanized anti-human CD16 antibody hCD16-M1 (SEQ ID NO:72). Residues 233-237 correspond to a linker (SEQ ID NO:21, underlined). Residues 238-265 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). Residues 266-278 of the first polypeptide chain correspond to a linker (SEQ ID NO:43). Residues 279-495 of the first polypeptide chain correspond to a “knob-bearing” IgG1 CH2-CH3 Domain (SEQ ID NO:51), in which the final residue is lysine. The second polypeptide chain of DART-3 has the amino acid sequence of SEQ ID NO:170: Residues 1-107 of the second polypeptide chain (SEQ ID NO:170) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of humanized anti-human CD16 antibody hCD16-M1 (SEQ ID NO:73). Residues 108-115 (double underlined) of the second polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-235 of the second polypeptide chain correspond to the VH Domain of vPalivizumab (SEQ ID NO:144). Residues 236-240 of the second polypeptide chain correspond to a linker (SEQ ID NO:21, underlined). Residues 241-268 of the second polypeptide chain correspond to a cysteine-containing K-coil (SEQ ID NO:32). The third polypeptide chain of the DART-3 has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The CD16 x HIV env Binding Molecule designated “DART-4” is a further illustrative CD16 x DA Binding Molecule. DART-4 is a bispecific diabody capable of binding CD16 and the HIV env protein. DART-4 is essentially the same as DART-1, except that it possesses the VH and VL Domains of the anti-HIV env protein antibody 7B2 (SEQ ID NO:140 and SEQ ID NO:141, respectively) instead of the VH and VL Domains of Trastuzumab (SEQ ID NO:124 and SEQ ID NO:125, respectively) that are present in DART-1. Thus, DART-4 is composed of three polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of the humanized anti-human CD16 antibody h3G8 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of 7B2 (and is thus immunospecific for an epitope of the HIV Env protein). The third polypeptide chain has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The three polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of the HIV env protein (see, e.g., The CD16 x RSV Binding Molecule designated “DART-5” is a further illustrative CD16 x DA Binding Molecule. DART-5 is a bispecific diabody capable of binding CD16 and the RSV glycoprotein F. DART-5 is essentially the same as DART-L, except that it possesses the VL and VH Domains of the anti-RSV glycoprotein F antibody vPalivizumab (SEQ ID NO:144 and SEQ ID NO:145, respectively) instead of the VH and VL Domains of CD19 mAb 1 (SEQ ID NO:138 and SEQ ID NO:139, respectively) that are present in DART-L. Thus, DART-5 is composed of three polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of the humanized anti-human CD16 antibody hCD16-M1 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of vPalivizumab (and is thus immunospecific for an epitope of the RSV glycoprotein F). The third polypeptide chain has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The three polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of RSV glycoprotein F (see, e.g., The CD16 x RSV Binding Molecule designated “DART-6” is a further illustrative CD16 x DA Binding Molecule. DART-6 is a bispecific diabody capable of binding CD16 and the RSV glycoprotein F. DART-6 is essentially the same as DART-M, except that it possesses the VL and VH Domains of the anti-RSV glycoprotein F antibody vPalivizumab (SEQ ID NO:144 and SEQ ID NO:145, respectively) instead of the VH and VL Domains of CD19 mAb 1 (SEQ ID NO:138 and SEQ ID NO:139, respectively) that are present in DART-M. Thus, DART-6 is composed of three polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of the optimized anti-human CD16 antibody hCD16-M1B (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of vPalivizumab (and is thus immunospecific for an epitope of the RSV glycoprotein F). The third polypeptide chain has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The three polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of RSV glycoprotein F (see, e.g., The CD16 x RSV Binding Molecule designated “DART-7” is a further illustrative CD16 x DA Binding Molecule. DART-7 is a bispecific diabody capable of binding CD16 and the RSV glycoprotein F. DART-7 is essentially the same as DART-N, except that it possesses the VL and VH Domains of the anti-RSV glycoprotein F antibody vPalivizumab (SEQ ID NO:144 and SEQ ID NO:145, respectively) instead of the VH and VL Domains of CD19 mAb 1 (SEQ ID NO:138 and SEQ ID NO:139, respectively) that are present in DART-N. Thus, DART-7 is composed of three polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of the optimized anti-human CD16 antibody hCD16-M1AB (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of vPalivizumab (and is thus immunospecific for an epitope of the RSV glycoprotein F). The third polypeptide chain has the amino acid sequence of the Common Diabody Polypeptide Chain (SEQ ID NO:153). The three polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of RSV glycoprotein F (see, e.g., The CD16 x HIV env Binding Molecule designated “DART-X” is a further illustrative CD16 x DA Binding Molecule. DART-X is a bispecific diabody capable of binding CD16 and the HIV env protein. DART-X is composed of two polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of the anti-human CD16 antibody CD16-M1 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of 7B2 (and is thus immunospecific for an epitope of the HIV env protein). The two polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of the HIV env protein (see, e.g., The first polypeptide chain of DART-X has the amino acid sequence of SEQ ID NO:171: Residues 1-113 of the first polypeptide chain (SEQ ID NO:171) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of 7B2 (SEQ ID NO:141). Residues 114-121 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 122-239 of the first polypeptide chain correspond to the VH Domain of the anti-human CD16 antibody CD16-M1 (SEQ ID NO:64). Residues 240-244 correspond to a linker (SEQ ID NO:21, underlined). Residues 245-272 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). The second polypeptide chain of DART-X has the amino acid sequence of SEQ ID NO:172: Residues 1-107 of the second polypeptide chain (SEQ ID NO:172) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of anti-human CD16 antibody CD16-M1 (SEQ ID NO:65). Residues 108-115 (double underlined) of the second polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-241 of the second polypeptide chain correspond to the VH Domain of 7B2 (SEQ ID NO:140). Residues 242-246 of the second polypeptide chain correspond to a linker (SEQ ID NO:21, underlined). Residues 247-274 of the second polypeptide chain correspond to a cysteine-containing K-coil (SEQ ID NO:32). The CD16 x HIV env Binding Molecule designated “DART-Y” is a further illustrative CD16 x DA Binding Molecule. DART-Y is a bispecific diabody capable of binding CD16 and the HIV env protein. DART-Y is composed of two polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of the anti-human CD16 antibody CD16-M2 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of 7B2 (and is thus immunospecific for an epitope of the HIV env protein). The two polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of the HIV env protein (see, e.g., The first polypeptide chain of DART-Y has the amino acid sequence of SEQ ID NO:173: Residues 1-113 of the first polypeptide chain (SEQ ID NO:173) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of 7B2 (SEQ ID NO:141). Residues 114-121 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 122-240 of the first polypeptide chain correspond to the VH Domain of the anti-human CD16 antibody CD16-M2 (SEQ ID NO:75). Residues 241-245 correspond to a linker (SEQ ID NO:21, underlined). Residues 246-273 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). The second polypeptide chain of DART-Y has the amino acid sequence of SEQ ID NO:174: Residues 1-107 of the second polypeptide chain (SEQ ID NO:174) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of anti-human CD16 antibody CD16-M2 (SEQ ID NO:76). Residues 108-115 (double underlined) of the second polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-241 of the second polypeptide chain correspond to the VH Domain of 7B2 (SEQ ID NO:140). Residues 242-246 of the second polypeptide chain correspond to a linker (SEQ ID NO:21, underlined). Residues 247-274 of the second polypeptide chain correspond to a cysteine-containing K-coil (SEQ ID NO:32). The CD16 x HIV env Binding Molecule designated “DART-0 is a further illustrative CD16 x DA Binding Molecule. DART-0 is a bispecific diabody capable of binding CD16 and the HIV env protein. DART-0 is essentially the same as DART-X, except that it possesses the VH and VL Domains of the humanized anti-human CD16 antibody h3G8 (SEQ ID NO:62 and SEQ ID NO:63, respectively) instead of the VH and VL Domains of anti-human CD16 antibody CD16-M1 (SEQ ID NO:64 and SEQ ID NO:65, respectively) that are present in the DART-X. Thus, DART-0 is composed of two polypeptide chains having one Binding Domain that comprises the VL and VH Domains of the humanized anti-human CD16 antibody h3G8 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of 7B2 (and is thus immunospecific for an epitope of the HIV Env protein). These polypeptide chains associate to form a covalently bonded DART® diabody composed of two polypeptide chains that possesses one Binding Domain immunospecific for the epitope of CD16 and one Binding Domain immunospecific for the epitope of the HIV env protein (see, e.g., The CD16 x HIV env Binding Molecule designated “DART-Z” is a further illustrative CD16 x DA Binding Molecule. DART-Z is a bispecific diabody capable of binding CD16 and the HIV env protein. DART-Z is essentially the same as DART-X, except that it possesses the VH and VL Domains of the anti-HIV env antibody A32 (SEQ ID NOs:142 and 143, respectively) rather than the VH and VL Domains of the anti-HIV env antibody 7B2, and it possesses the VH and VL Domains of the anti-human CD16A scFv 4-LS21 (U.S. Patent Publn. No. 2015/0218275): VH Domain of Anti-Human CD16A scFv 4-LS21 (SEQ ID NO:175): The CD16 x DA Binding Molecules of the present invention are further illustrated by Fc Domain-containing, bispecific or trispecific, CD16 x HIV env x HIV env Trivalent Molecules that comprise two Binding Domains capable of binding to an epitope of the HIV env protein and one Binding Domain capable of binding to an epitope of CD16. Such epitopes of the HIV env protein may be the same (as in the CD16 x HIV env x HIV env Trivalent Molecule “TRIDENT-A”) or they may be different (as in the CD16 x HIV env x HIV env Trivalent Molecule “TRIDENT-B”). The structures and sequences of illustrative Trivalent TRIDENT™ Molecules are summarized in Table 13, and are described in detail below. CD16 x HIV env x HIV env Trivalent Molecule designated “TRIDENT-A” is composed of four polypeptide chains and possesses one Binding Domain that comprises the VL and VH Domains of anti-human CD16 antibody hCD16-M1 (and is thus immunospecific for an epitope of CD16) and two Binding Domains that each comprise the VL and VH Domains of A32 (and is thus immunospecific for the epitope of the HIV env protein recognized by antibody A32). The four polypeptide chains associate to form a covalently bonded TRIDENT™ molecule capable of immunospecifically binding one or two copies of the HIV env protein epitope recognized by antibody A32, and an epitope of CD16 (see The first polypeptide chain of TRIDENT-A has the amino acid sequence of SEQ ID NO:177: Residues 1-110 of the first polypeptide chain (SEQ ID NO:177) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of Antibody A32 (SEQ ID NO:143). Residues 111-118 (double underlined) of the first polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 119-236 of the first polypeptide chain correspond to the VH Domain of anti-human CD16 antibody hCD16-M1 (SEQ ID NO:72). Residues 237-241 correspond to a linker (SEQ ID NO:21, underlined). Residues 242-269 of the first polypeptide chain correspond to a cysteine-containing E-coil (SEQ ID NO:31). Residues 270-282 of the first polypeptide chain correspond to a linker (SEQ ID NO:43). Residues 283-499 of the first polypeptide chain correspond to a “knob-bearing” IgG1 CH2-CH3 Domain (SEQ ID NO:51), in which the final residue is lysine. The second polypeptide chain of TRIDENT-A has the amino acid sequence of SEQ ID NO:178: Residues 1-107 of the second polypeptide chain (SEQ ID NO:178) of such illustrative CD16 x DA Binding Molecule correspond to the VL Domain of Antibody hCD16-M1 (SEQ ID NO:73). Residues 108-115 (double underlined) of the second polypeptide chain correspond to Linker 1 (GGGSGGGG; SEQ ID NO:16). Residues 116-238 of the second polypeptide chain correspond to the VH Domain of Antibody A32 (SEQ ID NO:142). Residues 239-243 of the second polypeptide chain correspond to a linker (SEQ ID NO:43). Residues 244-271 of the second polypeptide chain correspond to a cysteine-containing K-coil (SEQ ID NO:32). The third polypeptide chain of TRIDENT-A has the amino acid sequence of SEQ ID NO:179: Residues 1-123 of the third polypeptide chain (SEQ ID NO:179) correspond to the VH Domain of Antibody A32 (SEQ ID NO:142). Residues 124-221 of the third polypeptide chain correspond to a human IgG1 CH1 Domain (SEQ ID NO:3). Residues 222-236 of the third polypeptide chain correspond to an IgG Hinge Domain (SEQ ID NO:7, underlined). Residues 237-453 of the third polypeptide chain correspond to a “hole-bearing” IgG1 CH2-CH3 Domain (SEQ ID NO:53), in which the final residue is lysine. The fourth polypeptide chain of TRIDENT-A has the amino acid sequence of SEQ ID NO:180: Residues 1-110 of the fourth polypeptide chain (SEQ ID NO:180) correspond to the VL Domain of Antibody A32 (SEQ ID NO:143). Residues 111-217 of the fourth polypeptide chain correspond to a human CL Kappa Domain (SEQ ID NO:1). The illustrative CD16 x HIV env x HIV env Trivalent Molecule designated “TRIDENT-B” is also composed of four polypeptide chains. It possesses one Binding Domain that comprises the VL and VH Domains of anti-human CD16 antibody hCD16-M1 (and is thus immunospecific for an epitope of CD16), a Binding Domain that comprises the VL and VH Domains of A32 (and is thus immunospecific for the epitope of the HIV env protein recognized by antibody A32) and a further Binding Domain that comprises the VL and VH Domains of 7B2 (and is thus immunospecific for the epitope of the HIV env protein recognized by antibody 7B2). The four polypeptide chains associate to form a covalently bonded TRIDENT™ molecule capable of immunospecifically binding the epitope of the HIV env protein recognized by antibody A32 and/or the epitope of the HIV env protein recognized by antibody 7B2, and an epitope of CD16 (see The first polypeptide chain of TRIDENT-B is identical to the first polypeptide chain of TRIDENT-A (SEQ ID NO:177). The second polypeptide chain of TRIDENT-B is identical to the second polypeptide chain of TRIDENT-A (SEQ ID NO:178). The third polypeptide chain of TRIDENT-B has the amino acid sequence of SEQ ID NO:181: Residues 1-126 of the third polypeptide chain (SEQ ID NO:181) correspond to the VH Domain of Antibody 7B2 (SEQ ID NO:140). Residues 127-224 of the third polypeptide chain correspond to a human IgG1 CH1 Domain (SEQ ID NO:3). Residues 225-239 of the third polypeptide chain correspond to an IgG Hinge Domain (SEQ ID NO:7, underlined). Residues 240-456 of the third polypeptide chain correspond to a “hole-bearing” IgG1 CH2-CH3 Domain (SEQ ID NO:53), in which the final residue is lysine. The fourth polypeptide chain of TRIDENT-B has the amino acid sequence of SEQ ID NO:182: Residues 1-113 of the fourth polypeptide chain (SEQ ID NO:182) correspond to the VL Domain of Antibody 7B2 (SEQ ID NO:141). Residues 114-220 of the fourth polypeptide chain correspond to a human CL Kappa Domain (SEQ ID NO:1). The molecules of the present invention are most preferably produced through the recombinant expression of nucleic acid molecules that encode such polypeptides, as is well-known in the art. Polypeptides of the invention may be conveniently prepared using solid phase peptide synthesis (Merrifield, B. (1986) “ Antibodies may be made recombinantly and expressed using any method known in the art. Antibodies may be made recombinantly by first isolating the antibodies made from host animals, obtaining the gene sequence, and using the gene sequence to express the antibody recombinantly in host cells (e.g., CHO cells). Another method that may be employed is to express the antibody sequence in plants (e.g., tobacco) or transgenic milk. Suitable methods for expressing antibodies recombinantly in plants or milk have been disclosed (see, for example, Peeters et al. (2001) “ Vectors containing polynucleotides of interest (e.g., polynucleotides encoding the polypeptide chains of the binding molecules of the present invention) can be introduced into the host cell by any of a number of appropriate means, including electroporation, transfection employing calcium chloride, rubidium chloride, calcium phosphate, DEAE-dextran, or other substances; microprojectile bombardment; lipofection; and infection (e.g., where the vector is an infectious agent such as vaccinia virus). The choice of introducing vectors or polynucleotides will often depend on features of the host cell. Any host cell capable of overexpressing heterologous DNAs can be used for the purpose of expressing a polypeptide or protein of interest. Non-limiting examples of suitable mammalian host cells include but are not limited to COS, HeLa, and CHO cells. The invention includes polypeptides comprising an amino acid sequence of a binding molecule of this invention. The polypeptides of this invention can be made by procedures known in the art. The polypeptides can be produced by proteolytic or other degradation of the antibodies, by recombinant methods (i.e., single or fusion polypeptides) as described above or by chemical synthesis. Polypeptides of the antibodies, especially shorter polypeptides up to about 50 amino acids, are conveniently made by chemical synthesis. Methods of chemical synthesis are known in the art and are commercially available. The invention includes variants of the disclosed binding molecules, including functionally equivalent polypeptides that do not significantly affect the properties of such molecules as well as variants that have enhanced or decreased activity. Modification of polypeptides is routine practice in the art and need not be described in detail herein. Examples of modified polypeptides include polypeptides with conservative substitutions of amino acid residues, one or more deletions or additions of amino acids which do not significantly or deleteriously change the functional activity, or use of chemical analogs. Amino acid residues that can be conservatively substituted for one another include but are not limited to: glycine/alanine; serine/threonine; valine/isoleucine/leucine; asparagine/glutamine; aspartic acid/glutamic acid; lysine/arginine; and phenylalanine/tyrosine. These polypeptides also include glycosylated and non-glycosylated polypeptides, as well as polypeptides with other post-translational modifications, such as, for example, glycosylation with different sugars, acetylation, and phosphorylation. Preferably, the amino acid substitutions would be conservative, i.e., the substituted amino acid would possess similar chemical properties as that of the original amino acid. Such conservative substitutions are known in the art, and examples have been provided above. Amino acid modifications can range from changing or modifying one or more amino acids to complete redesign of a region, such as the Variable Domain. Changes in the Variable Domain can alter binding affinity and/or specificity. Other methods of modification include using coupling techniques known in the art, including, but not limited to, enzymatic means, oxidative substitution and chelation. Modifications can be used, for example, for attachment of labels for immunoassay, such as the attachment of radioactive moieties for radioimmunoassay. Modified polypeptides are made using established procedures in the art and can be screened using standard assays known in the art. In one embodiment, a fusion polypeptide is provided that comprises a Light Chain, a Heavy Chain or both a Light and Heavy Chain. In another embodiment, the fusion polypeptide contains a heterologous immunoglobulin constant region. In another embodiment, the fusion polypeptide contains a VH and a VL Domain of an antibody produced from a publicly-deposited hybridoma. For purposes of this invention, an antibody fusion protein contains polypeptide domains that enable the protein to immunospecifically bind both CD16 and a Disease Antigen, and which contains another amino acid sequence to which it is not attached in the native molecule, for example, a heterologous sequence or a homologous sequence from another region (e.g., a deimmunized albumin-binding domain, a Protein A recognition sequence, a peptide tag, etc.). The present invention particularly encompasses such binding molecules (e.g., antibodies, diabodies, trivalent binding molecules, etc.) conjugated to a diagnostic or therapeutic moiety. For diagnostic purposes, the binding molecules of the invention may be coupled to a detectable substance. Such binding molecules are useful for monitoring and/or prognosing the development or progression of a disease as part of a clinical testing procedure, such as determining the efficacy of a particular therapy. Examples of detectable substances include various enzymes (e.g., horseradish peroxidase, beta-galactosidase, etc.), prosthetic groups (e.g., avidin/biotin), fluorescent materials (e.g., umbelliferone, fluorescein, or phycoerythrin), luminescent materials (e.g., luminol), bioluminescent materials (e.g., luciferase or aequorin), radioactive materials (e.g., carbon-14, manganese-54, strontium-85 or zinc-65), positron emitting metals, and nonradioactive paramagnetic metal ions. The detectable substance may be coupled or conjugated either directly to the binding molecule or indirectly, through an intermediate (e.g., a linker) using techniques known in the art. For therapeutic purposes, the binding molecules of the invention may be conjugated to a therapeutic moiety such as a cytotoxin, (e.g., a cytostatic or cytocidal agent), a therapeutic agent or a radioactive metal ion, e.g., alpha-emitters. A cytotoxin or cytotoxic agent includes any agent that is detrimental to cells such as, for example, As discussed above, molecules capable of binding CD16 and a Disease Antigen are capable of mediating the redirected cell killing of a target cell (i.e., a cancer cell, or a pathogen-infected cell) that expresses such Disease Antigen on its cell surface. Such molecules may be used for therapeutic purposes, for example in subjects with cancer or an infection. Thus, binding molecules of the present invention have the ability to treat any disease or condition associated with or characterized by the expression of a Disease Antigen, particularly a Cancer Antigen or a Pathogen-Associated Antigen, on the surface of such target cell. Thus, without limitation, the binding molecules of the present invention may be employed in the treatment of cancer, particularly a cancer characterized by the expression of a Cancer Antigen. The binding molecules of the present invention may be employed in the treatment of infection, particularly an infection characterized by the expression of a Pathogen-Associated Antigen. In particular, the present invention encompasses such methods wherein the molecule capable of binding CD16 comprises an Epitope-Binding Domain of an antibody that is capable of binding CD16 and also comprises an Epitope-Binding Domain capable of binding a Disease Antigen (in particular a Cancer Antigen or a Pathogen-Associated Antigen) on the surface of a target cell so as to mediate the redirected killing of the target cell (for example, by mediating redirected cell killing (e.g., redirected T-cell or redirected NK-cell cytotoxicity)). In a specific embodiment, the molecule capable of binding CD16 and the Disease Antigen is a bispecific antibody, or the binding portions thereof, (including an scFv), a BiTe, a TandAb, and a CAR. In a specific embodiment, the molecule capable of binding CD16 and the Disease Antigen is a bispecific diabody. In a specific embodiment, the molecule capable of binding CD16 and the Disease Antigen is a trivalent binding molecule. As used herein, the terms: “providing a therapy” and “treating” refer to any administration of a composition that is associated with any indicia of beneficial or desired result, including, without limitation, any clinical result such as decreasing symptoms resulting from the disease, attenuating a symptom of infection (e.g., viral load, fever, pain, sepsis, etc.) a shrinking of the size of a tumor (in the cancer context, for example, a tumor of breast, gastric or prostate cancer), a retardation of cancer cell growth, a delaying of the onset, development or progression of metastasis, a decreasing of a symptom resulting from the disease, an increasing of the quality of life of the recipient subject, a decreasing of the dose of other medications being provided to treat a subject's disease, an enhancing of the effect of another medication such as via targeting and/or internalization, a delaying of the progression of the disease, and/or a prolonging of the survival of recipient subject. Subjects for treatment include animals, most preferably mammalian species such as non-primate (e.g., bovine, equine, feline, canine, rodent, etc.) or a primate (e.g., monkey such as, a cynomolgus monkey, human, etc.). In a preferred embodiment, the subject is a human. Exemplary disorders that may be treated by various embodiments of the present invention include, but are not limited to, proliferative disorders, cell proliferative disorders, and cancer (especially a cancer expressing a Cancer Antigen bound by a molecule capable of mediating redirected cell killing), pathogen-associated diseases (especially a chronic viral infection associated with expression of a Pathogen-Associated Antigen bound by a molecule capable of mediating redirected cell killing). In various embodiments, the invention encompasses methods and compositions for treatment, prevention or management of a disease or disorder in a subject, comprising administering to the subject a therapeutically effective amount the binding molecules of the present invention. Such molecules are particularly useful for the prevention, inhibition, reduction of growth, or regression of primary tumors, and metastasis of tumors, and for reducing pathogen load, or eliminating pathogen-infected cells. Although not intending to be bound by a particular mechanism of action, such molecules may mediate effector function against target cells, promote the activation of the immune system against target cells, cross-link cell-surface antigens and/or receptors on target cells and enhance apoptosis or negative growth regulatory signaling, or a combination thereof, resulting in clearance and/or reduction in the number of target cells. The cancers that may be treated by molecules of the present invention, and by the methods of the present invention, include, but are not limited to: an adrenal gland cancer, including but not limited to, a pheochromocytom or an adrenocortical carcinoma; an AIDS-associated cancer; an alveolar soft part sarcoma; an astrocytic tumor; a basal cancer; a bladder cancer, including but not limited to, a transitional cell carcinoma, a squamous cell cancer, an adenocarcinoma, or a carcinosarcoma; a bone and connective tissue sarcoma, such as but not limited to, a bone sarcoma, osteosarcoma, chondrosarcoma, Ewing's sarcoma, malignant giant cell tumor, fibrosarcoma of bone, chordoma, periosteal sarcoma, soft-tissue sarcomas, angiosarcoma (hemangiosarcoma), fibrosarcoma, Kaposi's sarcoma, leiomyosarcoma, liposarcoma, lymphangiosarcoma, neurilemmoma, rhabdomyosarcoma, or a synovial sarcoma; a brain cancer, including, but not limited to, a glioma, astrocytoma, brain stem glioma, ependymoma, oligodendroglioma, nonglial tumor, acoustic neurinoma, craniopharyngioma, medulloblastoma, meningioma, pineocytoma, pineoblastoma, or a primary brain lymphoma; a brain and spinal cord cancer; a breast cancer, including, but not limited to, an adenocarcinoma, lobular (small cell) carcinoma, intraductal carcinoma, medullary breast cancer, mucinous breast cancer, tubular breast cancer, papillary breast cancer, Paget's disease, or an inflammatory breast cancer; a carotid body tumor; a cervical cancer, including but not limited to, a squamous cell carcinoma, or a adenocarcinoma; a cholangiocarcinoma, including but not limited to, a papillary, nodular, or diffuse cholangiocarcinoma; a chondrosarcoma; a chordoma; a chromophobe renal cell carcinoma; a clear cell carcinoma; a colon cancer; a colorectal cancer; a cutaneous benign fibrous histiocytoma; a desmoplastic small round cell tumor; an ependymoma; an eye cancer, including, but not limited to, an ocular melanoma such as iris melanoma, choroidal melanoma, and cilliary body melanoma, and retinoblastoma; an esophageal cancer, including but not limited to, a squamous cancer, adenocarcinoma, adenoid cyctic carcinoma, mucoepidermoid carcinoma, adenosquamous carcinoma, sarcoma, melanoma, plasmacytoma, verrucous carcinoma, and an oat cell (small cell) carcinoma; a Ewing's tumor; an extraskeletal myxoid chondrosarcoma; a fibrogenesis imperfecta ossium; a fibrous dysplasia of the bone; a gallbladder or bile duct cancer, including but not limited to, an adenocarcinoma; a gastric cancer; a gestational trophoblastic disease; a germ cell tumor; a head and neck cancer; a hepatocellular carcinoma; Heavy Chain disease; an islet cell tumor; a Kaposi's sarcoma; a leukemia, including, but not limited to, an acute leukemia; acute lymphocytic leukemia; an acute myelocytic leukemia, such as, but not limited to, a myeloblastic, promyelocytic, myelomonocytic, monocytic, or erythroleukemia leukemia or a myelodysplastic syndrome; a chronic leukemia, such as but not limited to, a chronic myelocytic (granulocytic) leukemia, a chronic lymphocytic leukemia, a hairy cell leukemia; a lipoma/benign lipomatous tumor; a liposarcoma/malignant lipomatous tumor; a liver cancer, including but not limited to, a hepatocellular carcinoma, or a hepatoblastoma; a lymphoma, such as but not limited to, Hodgkin's disease; non-Hodgkin's disease; a lung cancer, including but not limited to, a non-small cell lung cancer, squamous cell carcinoma (epidermoid carcinoma), adenocarcinoma, large-cell carcinoma or a small-cell lung cancer; a medulloblastoma; a melanoma; a meningioma; a benign monoclonal gammopathy; a monoclonal gammopathy of undetermined significance; a multiple endocrine neoplasia; a multiple myeloma, such as but not limited to, a smoldering multiple myeloma, nonsecretory myeloma, osteosclerotic myeloma, plasma cell leukemia, solitary plasmacytoma and extramedullary plasmacytoma; a myelodysplastic syndrome; a neuroblastoma; a neuroendocrine tumor; an oral cancer, including but not limited to, a squamous cell carcinoma; an ovarian cancer; including, but not limited to, an ovarian epithelial carcinoma, borderline tumor, germ cell tumor, and stromal tumor; a pancreatic cancer, including but not limited to, an insulinoma, gastrinoma, glucagonoma, vipoma, somatostatin-secreting tumor, or a carcinoid or islet cell tumor; a parathyroid tumor; a pediatric cancer; a penal cancer; a peripheral nerve sheath tumor; a phaeochromocytoma; a pharynx cancer, including but not limited to, a squamous cell cancer, or a verrucous cancer; a pituitary cancer, including but not limited to, Cushing's disease, a prolactin-secreting tumor, acromegaly, or a diabetes insipius tumor; a prostate cancer, including but not limited to, an adenocarcinoma, leiomyosarcoma, or rhabdomyosarcoma; polycythemia vera; a posterious uveal melanoma; a rare hematologic disorder; a renal cancer, including but not limited to, an adenocarcinoma, hypernephroma, fibrosarcoma, a renal metastatic cancer, or a transitional cell cancer (renal pelvis and/or uterer); a rhabdoid tumor; a rhabdomysarcoma; a salivary gland cancer, including but not limited to, an adenocarcinoma, mucoepidermoid carcinoma, or an adenoidcystic carcinoma; a sarcoma; a skin cancer, including but not limited to, a basal cell carcinoma, a squamous cell carcinoma and melanoma, a superficial spreading melanoma, a nodular melanoma, a lentigo malignant melanoma, or an acral lentiginous melanoma; a soft-tissue sarcoma; a squamous cell cancer; a stomach cancer, including but not limited to, an adenocarcinoma, a fungating (polypoid), ulcerating, superficial spreading, diffusely spreading, or malignant lymphoma, liposarcoma, fibrosarcoma, and carcinosarcoma; a synovial sarcoma; a testicular cancer, including but not limited to, a germinal tumor, seminoma, anaplastic, classic (typical), spermatocytic, nonseminoma, embryonal carcinoma, teratoma carcinoma, or a choriocarcinoma (yolk-sac tumor); a thymic carcinoma; a thymoma; a thyroid cancer, such as but not limited to, papillary or follicular thyroid cancer, metastatic thyroid cancer, medullary thyroid cancer or anaplastic thyroid cancer; a uterine cancer, including but not limited to, an endometrial carcinoma or a uterine sarcoma; a vaginal cancer, including but not limited to, a squamous cell carcinoma, adenocarcinoma, or melanoma; a vulvar cancer, including but not limited to, a squamous cell carcinoma, melanoma, adenocarcinoma, basal cell carcinoma, sarcoma, or Paget's disease; a Waldenström's macroglobulinemia, or Wilms' tumor. In addition, cancers include myxosarcoma, osteogenic sarcoma, endotheliosarcoma, lymphangio-endotheliosarcoma, mesothelioma, synovioma, hemangioblastoma, epithelial carcinoma, cystadenocarcinoma, bronchogenic carcinoma, sweat gland carcinoma, sebaceous gland carcinoma, papillary carcinoma and papillary adenocarcinomas (for a review of such disorders, see Fishman et al., 1985, Medicine, 2d Ed., J.B. Lippincott Co., Philadelphia and Murphy et al., 1997, Informed Decisions: The Complete Book of Cancer Diagnosis, Treatment, and Recovery, Viking Penguin, Penguin Books U.S.A., Inc.). In particular, the binding molecules of the present invention may be used in the treatment of adrenal cancer, bladder cancer, breast cancer, colorectal cancer, gastric cancer, glioblastoma, kidney cancer, non-small-cell lung cancer, acute lymphocytic leukemia, acute myeloid leukemia, chronic lymphocytic leukemia, chronic myeloid leukemia, hairy cell leukemia, Burkett's lymphoma, diffuse large B cell lymphoma, follicular lymphoma, mantle cell lymphoma, marginal zone lymphoma, non-Hodgkin's lymphoma, small lymphocytic lymphoma, multiple myeloma, melanoma, ovarian cancer, pancreatic cancer, prostate cancer, skin cancer, renal cell carcinoma, testicular cancer, and uterine cancer. Pathogen-associated diseases that may be treated by the CD16 Binding Molecules of the present invention include chronic viral, bacterial, fungal and parasitic infections. Chronic infections that may be treated by the CD16 Binding Molecules of the present invention include Epstein Barr virus, Hepatitis A Virus (HAV); Hepatitis B Virus (HBV); Hepatitis C Virus (HCV); herpes viruses (e.g. HSV-1, HSV-2, CMV), Human Immunodeficiency Virus (HIV), Vesicular Stomatitis Virus (VSV), Bacilli, The present invention encompasses compositions comprising a molecule capable of binding CD16 and also capable of binding to a Disease Antigen. The compositions of the invention include bulk drug compositions useful in the manufacture of pharmaceutical compositions (e.g., impure or non-sterile compositions) and pharmaceutical compositions (i.e., compositions that are suitable for administration to a subject or patient) that can be used in the preparation of unit dosage forms. Such compositions comprise a prophylactically or therapeutically effective amount of a molecule capable of binding CD16 and also capable of binding to a Disease Antigen so as to be capable of mediating the redirected killing of a target cell (e.g., a cancer cell, a pathogen-infected cell, etc.), or a combination of such agents and a pharmaceutically acceptable carrier. Preferably, compositions of the invention comprise a prophylactically or therapeutically effective amount of the binding molecules of the present invention and a pharmaceutically acceptable carrier. In a preferred aspect, such compositions are substantially purified (i.e., substantially free from substances that limit its effect or produce undesired side effects). Various formulations of such compositions may be used for administration. In addition to the pharmacologically active agent(s), the compositions of the present invention may contain suitable pharmaceutically acceptable carriers comprising excipients and auxiliaries that are well-known in the art and are relatively inert substances that facilitate administration of a pharmacologically effective substance or which facilitate processing of the active compounds into preparations that can be used pharmaceutically for delivery to the site of action. For example, an excipient can give form or consistency, or act as a diluent. Suitable excipients include but are not limited to stabilizing agents, wetting and emulsifying agents, salts for varying osmolarity, encapsulating agents, buffers, and skin penetration enhancers. In a specific embodiment, the term “pharmaceutically acceptable” means approved by a regulatory agency of the Federal or a state government or listed in the U.S. Pharmacopeia or other generally recognized pharmacopeia for use in animals, and more particularly in humans. The term “carrier” refers to a diluent, adjuvant (e.g., Freund's adjuvant (complete and incomplete), excipient, or vehicle with which the therapeutic is administered. Generally, the ingredients of compositions of the invention are supplied either separately or mixed together in unit dosage form, for example, as a dry lyophilized powder or water free concentrate in a hermetically sealed container such as an ampoule or sachette indicating the quantity of active agent. Where the composition is to be administered by infusion, it can be dispensed with an infusion bottle containing sterile pharmaceutical grade water or saline. Where the composition is administered by injection, an ampoule of sterile water for injection or saline can be provided so that the ingredients may be mixed prior to administration. The invention also provides a pharmaceutical pack or kit comprising one or more containers filled with a binding molecule of the present invention, alone or with such pharmaceutically acceptable carrier. Additionally, one or more other prophylactic or therapeutic agents useful for the treatment of a disease can also be included in the pharmaceutical pack or kit. The invention also provides a pharmaceutical pack or kit comprising one or more containers filled with one or more of the ingredients of the pharmaceutical compositions of the invention. Optionally associated with such container(s) can be a notice in the form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceuticals or biological products, which notice reflects approval by the agency of manufacture, use or sale for human administration. The present invention provides kits that can be used in the above methods. A kit can comprise any of the binding molecules of the present invention. The kit can further comprise one or more other prophylactic and/or therapeutic agents useful for the treatment of cancer, in one or more containers. The compositions of the present invention may be provided for the treatment, prophylaxis, and amelioration of one or more symptoms associated with a disease, disorder or infection by administering to a subject an effective amount of a CD16 x DA pharmaceutical composition of the present invention. In a preferred aspect, such compositions are substantially purified (i.e., substantially free from substances that limit its effect or produce undesired side effects). In a specific embodiment, the subject is an animal, preferably a mammal such as non-primate (e.g., bovine, equine, feline, canine, rodent, etc.) or a primate (e.g., monkey such as, a cynomolgus monkey, human, etc.). In a preferred embodiment, the subject is a human. Methods of administering a CD16 x DA Binding Molecule or pharmaceutical composition of the invention include, but are not limited to, parenteral administration (e.g., intradermal, intramuscular, intraperitoneal, intravenous and subcutaneous), epidural, and mucosal (e.g., intranasal and oral routes). In a specific embodiment, the binding molecules of the present invention are administered intramuscularly, intravenously, or subcutaneously. The compositions may be administered by any convenient route, for example, by infusion or bolus injection, by absorption through epithelial or mucocutaneous linings (e.g., oral mucosa, rectal and intestinal mucosa, etc.) and may be administered together with other biologically active agents. Administration can be systemic or local. The invention also provides that preparations of the binding molecules of the present invention are packaged in a hermetically sealed container such as an ampoule or sachette indicating the quantity of the molecule. In one embodiment, such molecules are supplied as a dry sterilized lyophilized powder or water free concentrate in a hermetically sealed container and can be reconstituted, e.g., with water or saline to the appropriate concentration for administration to a subject. Preferably, the binding molecules of the present invention are supplied as a dry sterile lyophilized powder in a hermetically sealed container. The lyophilized preparations of the binding molecules of the present invention should be stored at between 2° C. and 8° C. in their original container and the molecules should be administered within 12 hours, preferably within 6 hours, within 5 hours, within 3 hours, or within 1 hour after being reconstituted. In an alternative embodiment, such molecules are supplied in liquid form in a hermetically sealed container indicating the quantity and concentration of the molecule, fusion protein, or conjugated molecule. Preferably, such binding molecules, when provided in liquid form, are supplied in a hermetically sealed container. The amount of such preparations of the invention that will be effective in the treatment, prevention or amelioration of one or more symptoms associated with a disorder can be determined by standard clinical techniques. The precise dose to be employed in the formulation will also depend on the route of administration, and the seriousness of the condition, and should be decided according to the judgment of the practitioner and each patient's circumstances. Effective doses may be extrapolated from dose-response curves derived from in vitro or animal model test systems. As used herein, an “effective amount” of a pharmaceutical composition is an amount sufficient to effect beneficial or desired results including, without limitation, clinical results such as decreasing symptoms resulting from the disease, attenuating a symptom of infection (e.g., viral load, fever, pain, sepsis, etc.) or a symptom of cancer (e.g., the proliferation, of cancer cells, tumor presence, tumor metastases, etc.), thereby increasing the quality of life of those suffering from the disease, decreasing the dose of other medications required to treat the disease, enhancing the effect of another medication such as via targeting and/or internalization, delaying the progression of the disease, and/or prolonging survival of individuals. When applied to an individual active ingredient, administered alone, the term refers to that ingredient alone. When applied to a combination, the term refers to combined amounts of the active ingredients that result in the therapeutic effect, whether administered in combination, serially, or simultaneously. An effective amount can be administered in one or more administrations. For purposes of this invention, an effective amount of drug, compound, or pharmaceutical composition is an amount sufficient: to kill and/or reduce the proliferation of cancer cells, and/or to eliminate, reduce and/or delay the development of metastasis from a primary site of cancer; or to reduce the proliferation of (or the effect of) an infectious pathogen and to reduce and/or delay the development of the pathogen-mediated disease, either directly or indirectly. In some embodiments, an effective amount of a drug, compound, or pharmaceutical composition may or may not be achieved in conjunction with another drug, compound, or pharmaceutical composition. Thus, an “effective amount” may be considered in the context of administering one or more chemotherapeutic agents, and a single agent may be considered to be given in an effective amount if, in conjunction with one or more other agents, a desirable result may be or is achieved. While individual needs vary, determination of optimal ranges of effective amounts of each component is within the skill of the art. For the binding molecules encompassed by the invention, the dosage administered to a patient is preferably determined based upon the body weight (kg) of the recipient subject. For the binding molecules encompassed by the invention, the dosage administered to a patient is typically from about 0.01 μg/kg to about 30 mg/kg or more of the subject's body weight. The dosage and frequency of administration of a binding molecule of the present invention may be reduced or altered by enhancing uptake and tissue penetration of the molecule by modifications such as, for example, lipidation. The dosage of a binding molecule of the invention administered to a patient may be calculated for use as a single agent therapy. Alternatively, the molecule may be used in combination with other therapeutic compositions and the dosage administered to a patient are lower than when said molecules are used as a single agent therapy. The pharmaceutical compositions of the invention may be administered locally to the area in need of treatment; this may be achieved by, for example, and not by way of limitation, local infusion, by injection, or by means of an implant, said implant being of a porous, non-porous, or gelatinous material, including membranes, such as SIALASTIC® membranes, or fibers. Preferably, when administering a molecule of the invention, care must be taken to use materials to which the molecule does not absorb. The compositions of the invention can be delivered in a vesicle, in particular a liposome (See Langer (1990) “ Treatment of a subject with a therapeutically or prophylactically effective amount of a binding molecule of the present invention can include a single treatment or, preferably, can include a series of treatments. In a preferred example, a subject is treated with a pharmaceutical composition of the invention for between about 1 to 10 weeks, preferably between 2 to 8 weeks, more preferably between about 3 to 7 weeks, and even more preferably for about 4, 5, or 6 weeks. The pharmaceutical compositions of the invention can be administered once a day with such administration occurring once a week, twice a week, once every two weeks, once a month, once every six weeks, once every two months, twice a year or once per year, etc. Alternatively, the pharmaceutical compositions of the invention can be administered twice a day with such administration occurring once a week, twice a week, once every two weeks, once a month, once every six weeks, once every two months, twice a year or once per year, etc. Alternatively, the pharmaceutical compositions of the invention can be administered three times a day with such administration occurring once a week, twice a week, once every two weeks, once a month, once every six weeks, once every two months, twice a year or once per year, etc. It will also be appreciated that the effective dosage of the molecules used for treatment may increase or decrease over the course of a particular treatment. Having now generally described the invention, the same will be more readily understood through reference to the following examples, which are provided by way of illustration and are not intended to be limiting of the present invention unless specified. As discussed above, a series of DART® Diabody CD16 x DA Binding Molecules were generated incorporating different murine, humanized, or optimized anti-CD16 binding domains and having a Binding Domain that is immunospecific for either: (1) the HER2/neu tumor antigen (Binding Domain from Trastuzumab), (2) a Binding Domain that is immunospecific for an HIV antigen (Binding Domain from A32 or 7B2), (3) the CD19 B-cell antigen (Binding Domain from CD19 mAb 1), or (4) a Binding Domain that is immunospecific for an RSV antigen (Binding Domain from Palivizumab) (Table 14). Competition studies were performed with the two chain diabodies (DART-X, DART-Y and DART-0) comprising murine antibody CD16-M1, murine antibody CD16-M2, humanized antibody 3G8 and two widely-available commercial antibodies: LNK16 and DJ130c (Abcam, etc.) A ProteOn analysis was conducted to assess the binding of test articles to CD16 molecules that had been immobilized on a chip. The CD16 molecules were first incubated with 5 nM CD16-Fc (human IgG2) Fusion captured, followed by incubation with 10 nM of a first test article (test article 1), followed by an incubation with 10 nM of a second test article (test article 2). Binding was detected using anti-human Fc F(ab′)2 fragments. This study revealed that the CD16-M1, CD16-M2, LNK16 and 3G8 binding domains each bound different epitopes of CD16. CD16-M2 was found to compete with antibody DJ130c for binding to CD16; DJ130c has been reported to bind CD16 at a site that is non-overlapping with CD16's Fc binding site (Tamm, A. et al. (1996) “ The ability of CD16-M1, DJ130c and LNK16 to bind CD16A in presence of IgG was evaluated. It was found that IgG presence inhibited binding by NK16, but that IgG presence did not inhibit binding between CD16 and CD16-M1 was not inhibited by the presence of IgG, whereas L. The 3G8 epitope has been reported to overlap with CD16's Fc binding site (Tamm, A. et al. (1996) J. Biol. Chem. 271(7):3659-3666). Increasing concentrations of human IgG (hIgG) descreased the binding of antibody LNK16 to CD16, thus indicating that the epitope recognized by LNK16 overlaps with CD16's Fc binding site; in contrast, binding by the CD16 Binding domains of DJ130c or CD16-M1 was substantially unaffected, thus indicating that the CD16 epitopes recognized by these molecules did not overlap with CD16's Fc binding site ( As discussed above, CD16A possesses two major allotypes, reflecting an 158F vs. 158V polymorphism. As shown in Table 15, CD16 Binding Domains of CD16-M1 and CD16-M2 were found to bind to both CD16 allotypes with similar high affinity (approximately 3-25-fold better than h3G8, which has a 10-fold lower affinity to the CD16 158F allotype, relative to the CD16 158V allotype. The binding domain of h3G8 was found to exhibit a fast off-rate. The analysis was conducted using a BIACORE® format in which the diabody molecules were captured on a goat (Fab′)2 anti-human Fc surface, and the human CD16A V/F molecules (labeled with Avitag) were passed over the captured diabodies (normalized; 1:1 binding fit). A ProteOn analysis was conducted to assess the binding of the CD16 Binding Domains of the constructed diabodies to CD16-His tagged molecules (R&D Systems) that had been immobilized on a chip. The CD16 molecules were immobilized via an anti-PentaHis antibody, followed by incubation incubation with the diabodies. The results of the CD16 binding studies are summarized in Table 16 (the CD16 Binding Domain of DART-Z derives from scFv 4-LS21, which is specific for CD16A). The results indicate that diabodies comprising CD16-M1 or CD16-M2 CD16 Binding Domains exhibited higher affinity to both alleles of CD16A and higher affinity than comparator molecules. Diabodies comprising hCD16-M2 (VH1) CD16 Binding Domains exhibited slightly better binding than diabodies comprising the hCD16-M2 (VH2) CD16 Binding Domains (compare DART-D vs. DART-E). In order to demonstrate the ability of the CD16 x DA Binding Molecules of the invention to bind CD16, whole blood of donor subjects was incubated in the presence of CD16-binding diabodies: DART-F and DART-G, an HIV x CD3 diabody (as a positive control for T-cell binding via its CD3 Binding Domain), an HIV x RSV diabody (as a negative control for all binding), and an h3G8 x RSV diabody comparator molecule. Following incubation, the cells were labled with: anti-CD56-allophycocyanin (CD56 APC, NK cell marker), anti-CD66b-fluorescein isothiocyanate (CD66b FITC, neutrophil cell marker) and anti-CD3-peridinin chlorophyll protein-Cy5.5 (CD3 PerCP Cy5.5, T-cell marker), and anti-humanFc-Phycoerythrin (αhFc PE) to detect diabody binding and the cell surface. The labeled cells were analyzed by flow cytometry gated on NK cells. The co-staining results of the investigation for a first subject are shown in Similar binding was observed using whole blood of a second donor subject. Lymphocyte binding (CD16A F/V) and Neutrophil binding (CD16B, allotype undetermined) was assessed essentially as described using DART-F (hCD16-M1), DART-G (hCD16-M1), and the h3G8 x RSV diabody comparator. The results of the investigation are shown in In order to demonstrate the ability of the CD16 x DA Binding Molecules of the invention to distinguish glycosylation allotypes NA1 and NA2 of CD16B, DART-F (hCD16-M1) and the h3G8 x RSV diabody comparator were incubated in the presence of whole blood and analyzed essentially as described above. The results show diabody staining of whole blood that had been gated on CD66b+ cells (i.e., neutrophils and eosinophils). In order to further evaluate the ability of the CD16 x DA Binding Molecules of the invention to bind leukocytes, the ability of CD16 Binding Domains of CD16-M1 to bind NK cells and granulocytes from whole blood was assessed. DART-C was incubated in the presence of whole blood leukocyte cells and analyzed essentially as described above. The results show diabody staining of whole blood that had been gated on NK cells (0.6% of leukocytres) or on granulocytes (49% of leukocytes). Since the amino acid sequences of human CD16, cynomolgus monkey CD16 and murine CD16 share varying degrees of homology ( The CD16 binding domain of h3G8 was found to be able to bind CD16a of cynomolgus monkey with somewhat reduced affinity relative to its binding to human CD16a. The CD16 binding domains of DART-C (hCD16-M1) and DART-D (hCD16-M2) were found to have low affinity to cynomolgus monkey CD16. None of the tested CD16 binding molecules were found to be capable of binding to murine CD16, commensurate with its lower homology to human CD16. In order to demonstrate the ability of the CD16 x DA Binding Molecules of the invention to mediate cell killing, the CD16 x HER2/neu Binding Molecules:
In order to further demonstrate the ability of the CD16 x DA Binding Molecules of the invention to mediate cell killing, the CD16 x HIV env Binding Molecules:
As indicated above, DART-F and DART-G are both Fc Domain-containing diabodies composed of three polypeptide chains. In order to demonstrate the ability of CD16 x DA Binding Molecules of the invention that lack Fc Domains to mediate cell killing, the CD16 x HIV env Binding Molecules:
As noted above, hCD16-M1 was found to have low affinity to cynomolgus monkey CD16 (cynoCD16). Random mutagenesis was used to introduce substitutions within the CDRL3 (Kabat positions 90-95) and CDRH3 (Kabat positions 96-100) Domains of hCD16-M1. The mutants were screened to identify clones having enhanced binding to non-human primate CD16 (e.g., cynoCD16) and that retained binding affinity binding to both alleles of human CD16. A variant designated “hCD16-M1A” having a mutated CDRH3, and a variant designated “hCD16-M1B” having a mutated CDRL3 were selected for further analysis. In addition, a third variant combining the CDRH3 and CDRL3 mutations was generated and designated “hCD16-M1AB.” The amino acid sequence of the CDRs and VH and VL Domains have been provided above (see, e.g., Table 8). Exemplary CD16 x DA Binding Molecules incorporating the optimized anti-CD16 binding domains: hCD16-M1A, hCD16-M1B, or hCD16-M1AB, and having an anti-HER2/neu Binding Domain were generated and designated DART-I, DART-J, and DART-K, respectively (see, Table 12 for summary and above for detailed description and full amino acid sequences). The ability of the exemplary CD16 x HER2/neu Binding Molecules comprising the optimized hCD16-M1A, hCD16-M1B, and hCD16-M1AB, to bind CD16 expressed on the surface of human ( The co-staining results of the investigation are shown in The ability of CD16 x DA Binding Molecules having optimized hCD16-M1A, hCD16-M1B, and hCD16-M1AB binding domains, to mediate redirected cell killing of JIMT-1-Luc target cells with human PBMCs (huPBMCs) or cynomolgus monkey PBMC (cynoPBMCs) effector cells from several donors was evaluated using two different redirected cell killing assays. Representative data from these studies are presented in These data show that, although the CD16 x DA Binding Molecule comprising the CD16 binding domain of hCD16-M1 (DART-C) binds cynoCD16 with apparent low affinity, it is capable of mediating redirected cell killing. In addition, CD16 x DA Binding Molecules comprising the optimized CD16 binding domains hCD16-M1A (DART-I), hCD16-M1B (DART-J), or hCD16-M1AB (DART-K) exhibited improved cytotoxicity with CynoPBMCs while exhibiting only a slight reduction in cytotoxicity with huPBMCs as compared to the same molecule comprising hCD16-M1 (DART-C). In further studies, exemplary CD16 x DA Binding Molecules having an anti-CD19 Binding Domain and incorporating the anti-CD16 binding domain of hCD16-M1, or the optimized anti-CD16 binding domain of hCD16-M1B or hCD16-M1AB, were generated and designated DART-L, DART-M, and DART-N, respectively (see, Table 12 for summary and above for detailed description and full amino acid sequences). Three additional molecules were generated and designated DART-5 (comprising hCD16-M1), DART-6 (comprising hCD16-M1A), and DART-7 (comprising hCD16-M1AB), in which the CD19 Binding Domain was replaced with an anti-RSV binding domain (see, Table 12 for summary and above for detailed description), such exemplary CD16 x RSV Binding Molecules are used below as negative controls for CD19 binding. The ability of: DART-L, DART-M, and DART-N; the control molecules: DART-5, DART-6, and DART-7; or the CD3 x CD19 DART® diabody duvortuxizumab (also known as MGD011; amino acid sequence found in WHO Drug Information, 2016, Proposed INN: List 116, 30(4):627-629) to mediate redirected cell killing of Raji-Luc target cells with human PBMCs (huPBMCs) effector cells (E:T=30:1) was evaluated in the LDH and LUM cell killing assays essentially as described below. Representative data from these studies are presented in The exemplary CD16 x DA Binding Molecules DART-L, DART-M, DART-N (having a binding site for the B-cell antigen CD19) and the negative controls: DART-5 and DART-6 (having a binding site for CD16 and a binding site for RSV) were evaluated for their ability to mediate autologous B-cell depletion in vitro. Briefly, PMBCs isolated from human or cynomolgus monkey were incubated in supplemented medium in the presence of increasing concentrations DART-L, DART-M, DART-N and the control molecules lacking a CD19 binding domain DART-5 and DART-6. B-cell levels were analyzed by flow cytometry (using CD3 for negative selection and CD20 as a B-cell marker; CD3/CD20+) at 72 hours and 96 hours post incubation for huPBMCs ( CD16 x DA Binding Molecules comprising the optimized CD16 binding domains hCD16-M1B (DART-M), or hCD16-M1AB (DART-N), mediated a larger reduction in B-cells, with both huPBMCs and cynoPBMCs as compared to the same molecule comprising hCD16-M1 (DART-L). However, higher concentrations were required to deplete B-cells from cynoPBMCs. This trend was observed for multiple PBMC donors. Minimal B-cell depletion was observed for the control molecules lacking a CD19 binding domain (DART-5 and DART-6). LDH redirected cell killing assay: These assays may be performed using the CytoTox 96® Non-Radioactive Cytotoxicity Assay Kit (Promega), or similar, that quantitatively measures LDH release essentially as described below. Target cells (e.g., tumor target cells) are resuspended at a density of 4×105cells/mL (or appropriate density to achieve the desired E:T ratio) in assay media (e.g., RPMI 1640 without phenol red, 10% FBS, 1% pen/strep) and preferably a viability of higher than 90% at assay initiation, and isolated purified effector cells (e.g., PBMC or NK cells purified from human or non-human primate (e.g., cynomolgus monkey) donor) suspended in the assay media at the appropriate density (e.g., 4×105cells/mL) to achieve an effector-to-target (E:T) cell ratio of 10:1, or 30:1 (or other desired E:T ratio) are used. An aliquot of target cell suspension (e.g., 50 μL, 20,000 cells), an aliquot of effector cell suspension (e.g., 100 μL, 200,000 cells for 10:1 E:T ratio), and an aliquot (e.g., 50 μL) of serially diluted test article (e.g., 5 fold, or 10 fold) are added to duplicate wells of a microtiter plate and incubated (37° C. with 5% CO2) for 24-96 hours, or longer if desired. At the end of the incubation an aliquot of lysis solution (e.g., 30 μL) is added and the plates are incubated for ˜10 minutes to completely lyse the target cells. The plates are then centrifuged to pellet the cell debris (e.g., 212 x g for 5 minutes) and an aliquot (e.g., 40 μL) of supernatant of is transferred from each well of the assay plate to a flat-bottom ELISA plate and an aliquot (e.g., 40 μL) of LDH substrate solution is added to each well. Plates are incubated for 10-20 minutes at room temperature in the dark and an aliquot (e.g., 40 μL) of stop solution (Promega Cat #G183A) is added. The optical density is measured at 490 nm within 1 hour on a Victor2 Multilabel plate reader (Perkin Elmer #1420-014), or similar. Specific cell lysis is calculated from optical density (OD) data using the following formula: wherein “AICC” is antibody-independent cellular cytotoxicity, “MR” is maximal release and “SR” is spontaneous release. The dose-response curves are generated using GraphPad Prism 6 software (or similar) by curve fitting the cytotoxicity values to the sigmoidal dose-response function. Luminescence (LUM) redirected cell killing assay: These assays may be performed using the Steady-Glo luciferase substrate (Promega), or a similar sustrate, and quantitatively measure celluar luciferase activity in living target cells engineered to express the luciferase (luc) reporter gene (e.g., JIMT-1-Luc, Raji-Luc cells) essentially as described below. The preparation and set up for these assays is essentially identical to the LDH assay described above. Following incubation, an aliquot (e.g., 100 μL) of culture medium is removed from each well and an aliquot (e.g., 100 μL) of Steady-Glo luciferase substrate ((Promega), or similar) is subsequently added to each well, followed by pipetting up/down several times for complete lysis of target cells. The plates are incubated at room temperature in the dark for 10 minutes and then luminescence intensity is measured using a VictorX4 Multilabel plate reader (Perkin Elmer #1420-014, or similar) with luminescence relative light unit (RLU) as the read-out. RLU is indicative of relative viability of the target cells. Dose-response curves are generated using GraphPad Prism 6 software (or similar) by curve fitting the RLU values to the sigmoidal dose-response function. All publications and patents mentioned in this specification are herein incorporated by reference to the same extent as if each individual publication or patent application was specifically and individually indicated to be incorporated by reference in its entirety. While the invention has been described in connection with specific embodiments thereof, it will be understood that it is capable of further modifications and this application is intended to cover any variations, uses, or adaptations of the invention following, in general, the principles of the invention and including such departures from the present disclosure as come within known or customary practice within the art to which the invention pertains and as may be applied to the essential features hereinbefore set forth. The present invention is directed to molecules (e.g., an antibody, a diabody, an scFv, an antibody, a TandAb, etc.) capable of binding an epitope of human CD16 (a “CD16 Binding Molecule”). The present invention is further directed to CD 16 Binding Molecules that are capable of binding an epitope of human CD16 and one or more epitope(s) of a Disease Antigen (“DA”) (e.g., a “CD16 x DA Binding Molecule”). The present invention is particularly directed to such CD16 x DA Binding Molecules that are antibodies, or that comprise an Epitope Binding Domain thereof, or are diabodies (including DART® diabodies), bispecific antibodies, TandAbs, other multispecific binding molecules (e.g., trivalent TRIDENT™ molecules), etc. The invention particularly concerns CD16 x DA Binding Molecules that are capable of binding a Disease Antigen that is a Cancer Antigen or a Pathogen-Associated Antigen in addition to being able to bind CD 16. The invention particularly concerns the use of such CD16 and CD16 x DA Binding Molecules in the treatment of cancer and pathogen-associated diseases. The present invention is also directed to pharmaceutical compositions that comprise such molecule(s). 1. A CD16 x Disease Antigen (CD16 x DA) Binding Molecule comprising a CD16 Binding Domain capable of binding an epitope of CD16 and also a Disease Antigen-Binding Domain capable of binding an epitope of a Disease Antigen, wherein said CD16 Binding Domain comprises one or more of:
(I) (A) a CDRH1 Domain comprising the amino acid sequence of SEQ ID NO:66;
(B) a CDRH2 Domain comprising the amino acid sequence of SEQ ID NO:67; (C) a CDRH3 Domain comprising the amino acid sequence of SEQ ID NO:68 or SEQ ID NO:60; (D) a CDRL1 Domain comprising the amino acid sequence of SEQ ID NO:69 or SEQ ID NO:74; (E) a CDRL2 Domain comprising the amino acid sequence of SEQ ID NO:70; and (F) a CDRL3 Domain comprising the amino acid sequence of SEQ ID NO:71 or SEQ ID NO:61; (II) (A) a CDRH1 Domain comprising the amino acid sequence of SEQ ID NO:77;
(B) a CDRH2 Domain comprising the amino acid sequence of SEQ ID NO:78; (C) a CDRH3 Domain comprising the amino acid sequence of SEQ ID NO:79; (D) a CDRL1 Domain comprising the amino acid sequence of SEQ ID NO:80; (E) a CDRL2 Domain comprising the amino acid sequence of SEQ ID NO:81; and (F) a CDRL3 Domain comprising the amino acid sequence of SEQ ID NO:82; (III) a VH Domain comprising the amino acid sequence of SEQ ID NO:72, SEQ ID NO:83, SEQ ID NO:84, or SEQ ID NO:58; (IV) a VL Domain comprising the amino acid sequence of SEQ ID NO:73, SEQ ID NO:85, or SEQ ID NO:59; (V) a VH Domain comprising the amino acid sequence of SEQ ID NO:72 and a VL Domain comprising the amino acid sequence of SEQ ID NO:73; (VI) a VH Domain comprising the amino acid sequence of SEQ ID NO:83 or SEQ ID NO:84 and a VL Domain comprising the amino acid sequence of SEQ ID NO:85; (VII) a VH Domain comprising the amino acid sequence of SEQ ID NO:58 and a VL Domain comprising the amino acid sequence of SEQ ID NO:73; (VIII) a VH Domain comprising the amino acid sequence of SEQ ID NO:72 and a VL Domain comprising the amino acid sequence of SEQ ID NO:59; and (IX) a VH Domain comprising the amino acid sequence of SEQ ID NO:58 and a VL Domain comprising the amino acid sequence of SEQ ID NO:59. 2. The CD16 x Disease Antigen Binding Molecule of 3. The CD16 x Disease Antigen Binding Molecule of any one of 4. The CD16 x Disease Antigen Binding Molecule of any one of (A) a CDRH1 Domain comprising the amino acid sequence of SEQ ID NO:66; (B) a CDRH2 Domain comprising the amino acid sequence of SEQ ID NO:67; (C) a CDRH3 Domain comprising the amino acid sequence of SEQ ID NO:68, or SEQ ID NO:60; (D) a CDRL1 Domain comprising the amino acid sequence of SEQ ID NO:69 or SEQ ID NO:74; (E) a CDRL2 Domain comprising the amino acid sequence of SEQ ID NO:70; and (F) a CDRL3 Domain comprising the amino acid sequence of SEQ ID NO:71, or SEQ ID NO:61. 5. The CD16 x Disease Antigen Binding Molecule of any one of (A) a VH Domain comprising the amino acid sequence of SEQ ID NO:72, or SEQ ID NO:58; (B) a VL Domain comprising the amino acid sequence of SEQ ID NO:73, or SEQ ID NO:59; (C) a VH Domain comprising the amino acid sequence of SEQ ID NO:72 and a VL Domain comprising the amino acid sequence of SEQ ID NO:73; (D) a VH Domain comprising the amino acid sequence of SEQ ID NO:58 and a VL Domain comprising the amino acid sequence of SEQ ID NO:73; (E) a VH Domain comprising the amino acid sequence of SEQ ID NO:72 and a VL Domain comprising the amino acid sequence of SEQ ID NO:59; or (F) a VH Domain comprising the amino acid sequence of SEQ ID NO:58 and a VL Domain comprising the amino acid sequence of SEQ ID NO:59. 6. The CD16 x Disease Antigen Binding Molecule of any one of (A) a CDRH1 Domain comprising the amino acid sequence of SEQ ID NO:77; (B) a CDRH2 Domain comprising the amino acid sequence of SEQ ID NO:78; (C) a CDRH3 Domain comprising the amino acid sequence of SEQ ID NO:79; (D) a CDRL1 Domain comprising the amino acid sequence of SEQ ID NO:80; (E) a CDRL2 Domain comprising the amino acid sequence of SEQ ID NO:81; and (F) a CDRL3 Domain comprising the amino acid sequence of SEQ ID NO:82. 7. The CD16 x Disease Antigen Binding Molecule of any one of (A) a VH Domain comprising the amino acid sequence of SEQ ID NO:83, or SEQ ID NO:84; (B) a VL Domain comprising the amino acid sequence of or SEQ ID NO:85; or (C) a VH Domain comprising the amino acid sequence of SEQ ID NO:83 or SEQ ID NO:84 and a VL Domain comprising the amino acid sequence of SEQ ID NO:85. 8. The CD16 x Disease Antigen Binding Molecule of any one of 9. The CD16 x Disease Antigen Binding Molecule of 10. The CD16 x Disease Antigen Binding Molecule of any one of 11. The CD16 x Disease Antigen Binding Molecule of any one of 12. The CD16 x Disease Antigen Binding Molecule of any one of 13. The CD16 x Disease Antigen Binding Molecule of 14. The CD16 x Disease Antigen Binding Molecule of 15. The CD16 x Disease Antigen Binding Molecule of any one of (A) a diabody, said diabody being a covalently bonded complex that comprises two, or three, four or five polypeptide chains; or (B) a trivalent binding molecule, said trivalent binding molecule being a covalently bonded complex that comprises three, four or five polypeptide chains, or (C) a bispecific antibody. 16. The CD16 x Disease Antigen Binding Molecule of 17. The CD16 x Disease Antigen Binding Molecule of 18. The CD16 x Disease Antigen Binding Molecule of (A) one or more amino acid modifications that reduces the affinity of the variant Fc Region for an FcγR; and/or (B) one or more amino acid modifications that enhances the serum half-life of the variant Fc Region. 19. The CD16 x Disease Antigen Binding Molecule of (A) said modifications that reduces the affinity of the variant Fc Region for an FcγR comprise the substitution of L234A; L235A; or L234A and L235A; and (B) said modifications that enhances the serum half-life of the variant Fc Region comprise the substitution of M252Y; M252Y and S254T; M252Y and T256E; M252Y, S254T and T256E; or K288D and H435K, wherein said numbering is that of the EU index as in Kabat. 20. A CD16 Binding Molecule, that comprises:
(A) a CDRH1 Domain comprising the amino acid sequence of SEQ ID NO:66; (B) a CDRH2 Domain comprising the amino acid sequence of SEQ ID NO:67; (C) a CDRH3 Domain comprising the amino acid sequence of SEQ ID NO:68, or SEQ ID NO:60; (D) a CDRL1 Domain comprising the amino acid sequence of SEQ ID NO:69 or SEQ ID NO:74; (E) a CDRL2 Domain comprising the amino acid sequence of SEQ ID NO:70; and (F) a CDRL3 Domain comprising the amino acid sequence of SEQ ID NO:71, or SEQ ID NO:61. 21. The CD16 Binding Molecule of (A) a VH Domain comprising the amino acid sequence of SEQ ID NO:72, or SEQ ID NO:58; (B) a VL Domain comprising the amino acid sequence of SEQ ID NO:73, or SEQ ID NO:59; (C) a VH Domain comprising the amino acid sequence of SEQ ID NO:72 and a VL Domain comprising the amino acid sequence of SEQ ID NO:73; (D) a VH Domain comprising the amino acid sequence of SEQ ID NO:58 and a VL Domain comprising the amino acid sequence of SEQ ID NO:73; (E) a VH Domain comprising the amino acid sequence of SEQ ID NO:72 and a VL Domain comprising the amino acid sequence of SEQ ID NO:59; or (F) a VH Domain comprising the amino acid sequence of SEQ ID NO:58 and a VL Domain comprising the amino acid sequence of SEQ ID NO:59. 22. A CD16 Binding Molecule that comprises:
(A) a CDRH1 Domain comprising the amino acid sequence of SEQ ID NO:77; (B) a CDRH2 Domain comprising the amino acid sequence of SEQ ID NO:78; (C) a CDRH3 Domain comprising the amino acid sequence of SEQ ID NO:79; (D) a CDRL1 Domain comprising the amino acid sequence of SEQ ID NO:80; (E) a CDRL2 Domain comprising the amino acid sequence of SEQ ID NO:81; and (F) a CDRL3 Domain comprising the amino acid sequence of SEQ ID NO:82. 23. The CD16 Binding Molecule of (A) a VH Domain comprising the amino acid sequence of SEQ ID NO:83 or SEQ ID NO:84; (B) a VL Domain comprising the amino acid sequence of or SEQ ID NO:85; or (C) a VH Domain comprising the amino acid sequence of SEQ ID NO:83 or SEQ ID NO:84 and a VL Domain comprising the amino acid sequence of SEQ ID NO:85. 24. The CD16 Binding Molecule of any one of 25. A pharmaceutical composition that comprises the CD16 x Disease Antigen Binding Molecule of any one of 26. A pharmaceutical composition that comprises the CD16 Binding Molecule, of any one of 27. Use of the pharmaceutical composition of 28. A method for the treatment of a disease characterized by the expression of said Disease Antigen, comprising administering to a subject in need thereof a therapeutically effective amount of the pharmaceutical composition of 29. The use of 30. The use of 31. The use or method of 32. The use or method of 33. The use or method of 34. The use of 36. The use or method of 37. The use or method of claim 35, wherein said Disease Antigen is an HIV env antigen.CROSS-REFERENCE TO RELATED APPLICATIONS
REFERENCE TO SEQUENCE LISTING
FIELD OF THE INVENTION
BACKGROUND OF THE INVENTION
SUMMARY OF THE INVENTION
BRIEF DESCRIPTION OF THE DRAWINGS
DETAILED DESCRIPTION OF THE INVENTION
I. Antibodies and Other Binding Molecules
A. Antibodies
1. General Structural Attributes of Antibodies
(a) Constant Domains
(i) Light Chain Constant Domain
RTVAAPSVFI FPPSDEQLKS GTASVVCLLN NFYPREAKVQ WKVDNALQSG NSQESVTEQD SKDSTYSLSS TLTLSKADYE KHKVYACEVT HQGLSSPVTK SFNRGEC QPKAAPSVTL FPPSSEELQA NKATLVCLIS DFYPGAVTVA WKADSSPVKA GVETTPSKQS NNKYAASSYL SLTPEQWKSH RSYSCQVTHE GSTVEKTVAP TECS (ii) Heavy Chain CH1 Domains
ASTKGPSVFP LAPSSKSTSG GTAALGCLVK DYFPEPVTVS WNSGALTSGV HTFPAVLQSS GLYSLSSVVT VPSSSLGTQT YICNVNHKPS NTKVDKRV ASTKGPSVFP LAPCSRSTSE STAALGCLVK DYFPEPVTVS WNSGALTSGV HTFPAVLQSS GLYSLSSVVT VPSSNFGTQT YTCNVDHKPS NTKVDKTV ASTKGPSVFP LAPCSRSTSG GTAALGCLVK DYFPEPVTVS WNSGALTSGV HTFPAVLQSS GLYSLSSVVT VPSSSLGTQT YTCNVNHKPS NTKVDKRV ASTKGPSVFP LAPCSRSTSE STAALGCLVK DYFPEPVTVS WNSGALTSGV HTFPAVLQSS GLYSLSSVVT VPSSSLGTKT YTCNVDHKPS NTKVDKRV (b) Heavy Chain Hinge Regions
EPKSCDKTHTCPPCP. ERKCCVECPPCP. ELKTPLGDTT HTCPRCPEPK SCDTPPPCPR CPEPKSCDTP PPCPRCPEPK SCDTPPPCPR CP. ESKYGPPCPSCP.
As described herein, an IgG4 Hinge Domain may comprise a stabilizing mutation such as the S228P substitution. The amino acid sequence of an exemplary S228P-stabilized human IgG4 Hinge Domain is (SEQ ID NO:11):
ESKYGPPCPPCP. (c) Heavy Chain CH2 and CH3 Domains
231 240 250 260 270 APELLGGPSV FLFPPKPKDT LMISRTPEVT CVVVDVSHED 280 290 300 310 PEVKFNWYVD GVEVHNAKTK PREEQYNSTY RVVSVLTVLH 320 330 340 350 QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT 360 370 380 390 LPPSREEMTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN 400 410 420 430 YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE 440 447 ALHNHYTQKS LSLSPGX
as numbered by the EU index as set forth in Kabat, wherein X is lysine (K) or is absent.
231 240 250 260 270 APPVA-GPSV FLFPPKPKDT LMISRTPEVT CVVVDVSHED 280 290 300 310 PEVQFNWYVD GVEVHNAKTK PREEQFNSTF RVVSVLTVVH 320 330 340 350 QDWLNGKEYK CKVSNKGLPA PIEKTISKTK GQPREPQVYT 360 370 380 390 LPPSREEMTK NQVSLTCLVK GFYPSDISVE WESNGQPENN 400 410 420 430 YKTTPPMLDS DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE 440 447 ALHNHYTQKS LSLSPGX
as numbered by the EU index as set forth in Kabat, wherein X is lysine (K) or is absent.
231 240 250 260 270 APELLGGPSV FLFPPKPKDT LMISRTPEVT CVVVDVSHED 280 290 300 310 PEVQFKWYVD GVEVHNAKTK PREEQYNSTF RVVSVLTVLH 320 330 340 350 QDWLNGKEYK CKVSNKALPA PIEKTISKTK GQPREPQVYT 360 370 380 390 LPPSREEMTK NQVSLTCLVK GFYPSDIAVE WESSGQPENN 400 410 420 430 YNTTPPMLDS DGSFFLYSKL TVDKSRWQQG NIFSCSVMHE 440 447 ALHNRFTQKS LSLSPGX
as numbered by the EU index as set forth in Kabat, wherein X is lysine (K) or is absent.
231 240 250 260 270 APEFLGGPSV FLFPPKPKDT LMISRTPEVT CVVVDVSQED 280 290 300 310 PEVQFNWYVD GVEVHNAKTK PREEQFNSTY RVVSVLTVLH 320 330 340 350 QDWLNGKEYK CKVSNKGLPS SIEKTISKAK GQPREPQVYT 360 370 380 390 LPPSQEEMTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN 400 410 420 430 YKTTPPVLDS DGSFFLYSRL TVDKSRWQEG NVFSCSVMHE 440 447 ALHNHYTQKS LSLSLGX
as numbered by the EU index as set forth in Kabat, wherein X is lysine (K) or is absent.
(d) Variable Domains
2. Humanization of Antibodies
B. Bispecific Antibodies
C. Chimeric Antigen Receptors
II. Bispecific Diabodies
A. Diabodies Lacking Fc Domains
GGGS GGGG. LAEAKVLANR ELDKYGVSDY YKNLIDNAKS AEGVKALIDE ILAALP. (SEQ ID NO: 34) LAEAKVLANR ELDKYGVSDY YKNLID66NAKS70 A71EGVKALIDE ILAALP,
or the amino acid sequence:
(SEQ ID NO: 35) LAEAKVLANR ELDKYGVSDY YKNA64A65NNAKT VEGVKALIA79E ILAALP,
or the amino acid sequence:
(SEQ ID NO: 36) LAEAKVLANR ELDKYGVSDY YKNLIS66NAKS70 VEGVKALIA79E ILAALP,
are particularly preferred as such deimmunized ABD exhibit substantially wild-type binding while providing attenuated MHC class II binding. Thus, the first polypeptide chain of such a diabody having an ABD contains a third linker (Linker 3) preferably positioned C-terminally to the E-coil (or K-coil) Domain of such polypeptide chain so as to intervene between the E-coil (or K-coil) Domain and the ABD (which is preferably a deimmunized ABD). A preferred sequence for such Linker 3 is SEQ ID NO:18:
GGGS. B. Diabodies Comprising Fc Domains
(SEQ ID NO: 29: EVAALEK-EVAALEK-EVAALEK-EVAALEK or SEQ ID NO: 31: EVAACEK-EVAALEK-EVAALEK-EVAALEK); and the “K-coil” domains (SEQ ID NO: 30: KVAALKE-KVAALKE-KVAALKE-KVAALKE or SEQ ID NO: 32: KVAACKE-KVAALKE-KVAALKE-KVAALKE).
A representative coil domain containing four-chain diabody is shown in Bispecific 2ndChain NH2-VL2-VH1-HPD-COOH 1sChain NH2-VL1-VH2-HPD-CH2-CH3-COOH 1stChain NH2-VL1-VH2-HPD-CH2-CH3-COOH 2ndChain NH2-VL2-VH1-HPD-COOH Tetraspecific 2ndChain NH2-VL2-VH1-HPD-COOH 1stChain NH2-VL1-VH2-HPD-CH2-CH3-COOH 3rdChain NH2-VL3-VH4-HPD-CH2-CH3-COOH 4thChain NH2-VL4-VH3-HPD-COOH HPD = Heterodimer-Promoting Domain First 3rdChain NH2-CH2-CH3-COOH Orientation 1stChain NH2-VL1-VH2-HPD-CH2-CH3-COOH 2ndChain NH2-VL2-VH1-HPD-COOH Second 3rdChain NH2-CH2-CH3-COOH Orientation 1stChain NH2-CH2-CH3-VL1-VH2-HPD-COOH 2ndChain NH2-VL2-VH1-HPD-COOH HPD = Heterodimer-Promoting Domain Bispecific 2ndChain NH2-VL1-CL-COOH (2 × 2) 1stChain NH2-VH1-CH1-CH2-CH3-COOH 3rdChain NH2-VH1-CH1-CH2-CH3- VL2-VH2-HPD-COOH 5ndChain NH2-VL1-CL-COOH 4tthChain NH2-VL2-VH2-HPD-COOH Bispecific 2ndChain NH2-VL1-CL-COOH (3 × 1) 1stChain NH2-VH1-CH1-CH2-CH3-COOH 3rdChain NH2-VH1-CH1-CH2-CH3- VL1-VH2-HPD-COOH 5ndChain NH2-VL1-CL-COOH 4thChain NH2-VL2-VH1-HPD-COOH Trispecific 2ndChain NH2-VL1-CL-COOH (2 × 1 × 1) 1stChain NH2-VH1-CH1-CH2-CH3-COOH 3rdChain NH2-VH1-CH1-CH2-CH3- VL2-VH3-HPD-COOH 5ndChain NH2-VL1-CL-COOH 4thChain NH2-VL3-VH2-HPD-COOH HPD = Heterodimer-Promoting Domain Single-Site Variations F243L R292G D270E R292P Y300L P396L Double-SiteVariations F243L and F243L and F243L and R292P and R292P Y300L P396L Y300L D270E and R292P and P396L and P247L and P396L V305I Q419H N421K R292P and Y300L and R255L and R292P and P396L P396L P396L P305I K392T and P396L Triple-Site Variations F243L, P247L and N421K P247L, D270E and N421K F243L, R292P and Y300L R255L, D270E and P396L F243L, R292P and V305I D270E, G316D and R416G F243L, R292P and P396L D270E, K392T and P396L F243L, Y300L and P396L D270E, P396L and Q419H V284M, R292L and K370N R292P, Y300L and P396L Quadruple-Site Variations L234F, F243L, R292P and Y300L F243L, P247L, D270E and N421K L234F, F243L, R292P and Y300L F243L, R255L, D270E and P396L L235I, F243L, R292P and Y300L F243L, D270E, G316D and R416G L235Q, F243L, R292P and Y300L F243L, D270E, K392T and P396L P247L, D270E, Y300L and N421K F243L, R292P, Y300L, and P396L R255L, D270E, R292G and P396L F243L, R292P, V305I and P396L R255L, D270E, Y300L and P396L F243L, D270E, P396L and Q419H D270E, G316D, P396L and R416G Quintuple-SiteVariations L235V, F243L, R292P, F243L, R292P, V305I, Y300L and P396L Y300L and P396L L235P, F243L, R292P, Y300L and P396L †numbering is according to the EU index as in Kabat APEAAGGPSV FLFPPKPKDT LMISRTPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT LPPSREEMTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE ALHNHYTQKS LSLSPGX APELLGGPSV FLFPPKPKDT LMISRTPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT LPPSREEMTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE ALHNHYTQKS LCLSPGX
wherein X is lysine (K) or is absent.
APEAAGGPSV FLFPPKPKDT LMISRTPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT LPPSREEMTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE ALHNHYTQKS LCLSPGX
wherein X is lysine (K) or is absent.
APELLGGPSV FLFPPKPKDT LYITREPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT LPPSREEMTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE ALHNHYTQKS LSLSPGX APEAAGGPSV FLFPPKPKDT LYITREPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT LPPSREEMTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE ALHNHYTQKS LSLSPGX APEAAGGPSV FLFPPKPKDT LYITREPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT LPPSREEMTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE ALHNHYTQKS LCLSPGX
wherein X is lysine (K) or is absent.
APEAAGGPSV FLFPPKPKDT LMISRTPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT LPPSREEMTK NQVSLWCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE ALHNHYTQKS LSLSPGX APEAAGGPSV FLFPPKPKDT LYITREPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT LPPSREEMTK NQVSLWCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSTWQQG NVFSCSVMHE ALHNHYTQKS LSLSPGX APEAAGGPSV FLFPPKPKDT LMISRTPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT LPPSREEMTK NQVSLSCAVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLVSKL TVDKSRWQQG NVFSCSVMHE ALHNRYTQKS LSLSPGX APEAAGGPSV FLFPPKPKDT LYITREPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT LPPSREEMTK NQVSLSCAVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLVSKL TVDKSRWQQG NVFSCSVMHE ALHNRYTQKS LSLSPGX APEFLGGPSV FLFPPKPKDT LYITREPEVT CVVVDVSQED PEVQFNWYVD GVEVHNAKTK PREEQFNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKGLPS SIEKTISKAK GQPREPQVYT LPPSQEEMTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSRL TVDKSRWQEG NVFSCSVMHE ALHNHYTQKS LSLSLGX APEFLGGPSV FLFPPKPKDT LYITREPEVT CVVVDVSQED PEVQFNWYVD GVEVHNAKTK PREEQFNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKGLPS SIEKTISKAK GQPREPQVYT LPPSQEEMTK NQVSLWCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSRL TVDKSRWQEG NVFSCSVMHE ALHNHYTQKS LSLSLGX APEFLGGPSV FLFPPKPKDT LYITREPEVT CVVVDVSQED PEVQFNWYVD GVEVHNAKTK PREEQFNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKGLPS SIEKTISKAK GQPREPQVYT LPPSQEEMTK NQVSLSCAVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLVSRL TVDKSRWQEG NVFSCSVMHE ALHNRYTQKS LSLSLGX III. Trivalent Binding Molecules Containing Fc Domains
GGGSGGGG.
Other Domains of the trivalent binding molecules may be separated by one or more intervening spacer peptides (Linkers), optionally comprising a cysteine residue. In particular, as provided above, such Linkers will typically be incorporated between Variable Domains (i.e., VH or VL) and peptide Heterodimer-Promoting Domains (e.g., an E-coil or K-coil) and between such peptide Heterodimer-Promoting Domains (e.g., an E-coil or K-coil) and CH2-CH3 Domains. Exemplary linkers useful for the generation of trivalent binding molecules are provided above and are also provided in PCT Application Nos: PCT/US15/33081; and PCT/US15/33076. Thus, the first and second polypeptide chains of such trivalent binding molecules associate together to form a VL1/VH1 Binding Domain capable of binding a First Epitope, as well as a VL2/VH2 Binding Domain that is capable of binding a Second Epitope. The third and fourth polypeptide chains of such trivalent binding molecules associate together to form a VL3/VH3 Binding Domain that is capable of binding a Third Epitope.
(SEQ ID NO: 41) GGGGSGGGGSGGGGS. Four Chain 2nd Chain NH2-VL2-VH1-HPD-COOH 1st 1st Chain NH2-VL1-VH2-HPD-CH2-CH3-COOH Orientation 3rdChain NH2-VH3-CH1-CH2-CH3-COOH 2ndChain NH2-VL3-CL-COOH Four Chain 2ndChain NH2-VL2-VH1-HPD-COOH 2nd 1stChain NH2-CH2-CH3-VL1-VH2-HPD-COOH Orientation 3rdChain NH2-VH3-CH1-CH2-CH3-COOH 2ndChain NH2-VL3-CL-COOH Three Chain 2nd Chain NH2-VL2-VH1-HPD-COOH 1st 1stChain NH2-VL1-VH2-HPD-CH2-CH3-COOH Orientation 3rdChain NH2-VL3-VH3-HPD-CH2-CH3-COOH Three Chain 2ndChain NH2-VL2-VH1-HPD-COOH 2nd 1st Chain NH2-CH2-CH3-VL1-VH2-HPD-COOH Orientation 3rdChain NH2-VL3-VH3-HPD-CH2-CH3-COOH HPD = Heterodimer-Promoting Domain IV. Embodiments of the Invention
Number of Epitopes Recognized by Exemplary CD16 × DA Binding Molecules of the Invention Possessing Two, Three or Four Epitope Binding Domains That Are Capable of Mediating the Redirected Killing of a Target Cell Number Number Number Number of of of Total of Epitopes Epitopes Epitopes Number Number Epitopes of of of of of of 2nd 3rd Non-CD16 Binding CD16 1stDisease Disease Disease Cell Surface Domains Epitope(s) Antigen Antigen Antigen Molecule 2 1 1 0 0 0 3 1 1 1 0 0 3 1 1 0 0 1 3 1 2 0 0 0 3 2 1 0 0 0 4 1 1 1 0 1 4 1 1 1 1 0 4 1 2 0 0 1 4 1 2 1 0 0 4 2 1 1 0 0 4 2 1 0 0 1 V. Exemplary Binding Molecules
A. Exemplary Anti-Human CD16 Antibodies
1. CD16-M1 and CD16-M2 and Their Humanized Derivatives hCD16-M1 and hCD16-M2
(a) Anti-Human CD16 Monoclonal Antibody CD16-M1
EVKLVESGGT LVKPGGSLKL SCAASGFTFN NYGMSWVRQT PEKRLEWVATISGGGSYTFYPDSVKGRFTI SRDNAKNSLY LQMSSLRSED TALYYCIRQSARAPEPYWGQ GTLVTVSS DIVMTQSQKF MSTSVGDRVS VTCKASQNVGTHVAWYQQKS GQSPKSLLYSASYRYSGVPD RFSGSGSGTD FTLTISNVQS EDLAEYFCQQYKSYPLTFGA GTKLELK CDRs of Anti-Human CD16-Monoclonal Antibody CD16-M1 CDR Sequence SEQ ID NO CDRH1 NYGMS SEQ ID NO: 66 CDRH2 TISGGGSYTFYPDSVKG SEQ ID NO: 67 CDRH3 QSARAPEPY SEQ ID NO: 68 CDRL1 KASQNVGTHVA SEQ ID NO: 69 CDRL2 SASYRYS SEQ ID NO: 70 CDRL3 QQYKSYPLT SEQ ID NO: 71 EVQLVESGGG LVKPGGSLRL SCAASGFTFS NYGMSWVRQA PGKGLEWVATISGGGSYTFYPDSVKGRFTI SRDNAKNSLY LQMNSLRTED TALYYCVRQSARAPEPYWGQ GTLVTVSS DIQMTQSPSF LSASVGDRVT ITCRASQNVGTHVAWYQQKP GKAPKSLLYSASYRYSGVPS RFSGSGSGTD FTLTISSLQS EDIATYYCQQYKSYPLTFGQ GTKLEIK EVQLVESGGG LVKPGGSLRL SCAASGFTFS NYGMSWVRQA PGKGLEWVAT ISGGGSYTFY PDSVKGRFTI SRDNAKNSLY LQMNSLRTED TALYYCVRQS ANSPVPYWGQ GTLVTVSS DIQMTQSPSF LSASVGDRVT ITCRASQNVG THVAWYQQKP GKAPKSLLYS ASYRYSGVPS RFSGSGSGTD FTLTISSLQS EDIATYYCQD YTNYPLTFGQ GTKLEIK CDRs of Anti-Human CD16-Monoclonal Antibody hCD16-M1 CDR Sequence SEQ ID NO CDRH1 NYGMS SEQ ID NO: 66 CDRH2 TISGGGSYTFYPDSVKG SEQ ID NO: 67 CDRH3 QSARAPEPY SEQ ID NO: 68 CDRL1 RASQNVGTHVA SEQ ID NO: 74 CDRL2 SASYRYS SEQ ID NO: 70 CDRL3 QQYKSYPLT SEQ ID NO: 71 hCD16-M1A CDRH1 NYGMS SEQ ID NO: 66 CDRH2 TISGGGSYTFYPDSVKG SEQ ID NO: 67 CDRH3 QSANSPVPY SEQ ID NO: 60 CDRL1 RASQNVGTHVA SEQ ID NO: 74 CDRL2 SASYRYS SEQ ID NO: 70 CDRL3 QQYKSYPLT SEQ ID NO: 71 hCD16-M1B CDRH1 NYGMS SEQ ID NO: 66 CDRH2 TISGGGSYTFYPDSVKG SEQ ID NO: 67 CDRH3 QSARAPEPY SEQ ID NO: 68 CDRL1 RASQNVGTHVA SEQ ID NO: 74 CDRL2 SASYRYS SEQ ID NO: 70 CDRL3 QDYTNYPLT SEQ ID NO: 61 hCD16-M1AB CDRH1 NYGMS SEQ ID NO: 66 CDRH2 TISGGGSYTFYPDSVKG SEQ ID NO: 67 CDRH3 QSANSPVPY SEQ ID NO: 60 CDRLI RASQNVGTHVA SEQ ID NO: 74 CDRL2 SASYRYS SEQ ID NO: 70 CDRL3 QDYTNYPLT SEQ ID NO: 61 (b) Anti-Human CD16 Monoclonal Antibody CD16-M2
EVQLQQSGPE LVKPGASVKM SCKASGYTFT SSAMHWVKKN PGQGLEWIGYINHYNDGIKYNERFKGKATL TSDKSSSTAY MELSSLTSED SAVYYCATGYRYASWFASWG QGTLVTVSS DILLTQSPAI LSVSPGERVS FSCRASQNIGTSIHWYQQRT DGSPRLLIKSVSESISGIPS RFSGSGSGTD FTLTINGVES GDISDYYCQQSNSWPLTFGA GTKLELK CDRs of Anti-Human CD16 Monoclonal Antibody CD16-M2 CDR Sequence SEQ ID NO CDRH1 SSAMH SEQ ID NO: 77 CDRH2 YINHYNDGIKYNERFKG SEQ ID NO: 78 CDRH3 GYRYASWFAS SEQ ID NO: 79 CDRL1 RASQNIGTSIH SEQ ID NO: 80 CDRL2 SVSESIS SEQ ID NO: 81 CDRL3 QQSNSWPLT SEQ ID NO: 82 QVQLVQSGAE VKKPGASVKV SCKASGYTFT SSAMHWVRQA PGQGLEWMGYINHYNDGIKY NERFKGRVTI TADKSTSTAY MELSSLRSED TAVYYCATGYRYASWFASWG QGTLVTVSS QVQLVQSGAE VKKPGASVKV SCKASGYTFT SSAMHWVRQA PGQGLEWMGYINHYNDGIKY NERFKGRVTI TADKSTSTAY MELSSLRSED TAVYYCAGYRYASWFASWG QGTLVTVSS EIVLTQSPAT LSVSPGERAT LSCRASQNIG TSIHWYQQKP DQSPKLLIKSVSESISGVPS RFSGSGSGTD FTLTINSLEA EDFATYYCQQ SNSWPLTFGQ GTKLEIK CDRs of Anti-Human CD16 Monoclonal Antibody hCD16-M2 CDR Sequence SEQ ID NO CDRH1 SSAMH SEQ ID NO: 77 CDRH2 YINHYNDGIKYNERFKG SEQ ID NO: 78 CDRH3 GYRYASWFAS SEQ ID NO: 79 CDRL1 RASQNIGTSIH SEQ ID NO: 80 CDRL2 SVSESIS SEQ ID NO: 81 CDRL3 QQSNSWPLT SEQ ID NO: 82 B. Exemplary Antibodies that Bind to the Cell Surface of Effector Cells
1. Exemplary Anti-NKG2D Antibodies
EVQLVESGGG VVQPGGSLRL SCAASGFTFS SYGMHWVRQA PGKGLEWVAFIRYDGSNKYY ADSVKGRFTI SRDNSKNTKY LQMNSLRAED TAVYYCAKDRFGYYLDYWGQ GTLVTVSS QPVLTQPSSV SVAPGETARI PCGGDDIETK SVHWYQQKPG QAPVLVIYDDDDRPSGIPER FFGSNSGNTA TLSISRVEAG DEADYYCQVW DDNNDEWVFG GGTQLTVL QVQLVESGGG LVKPGGSLRL SCAASGFTFS SYGMHWVRQA PGKGLEWVAFIRYDGSNKYY ADSVKGRFTI SRDNSKNTLY LQMNSLRAED TAVYYCAKDRGLGDGTYFDY WGQGTTVTVS S QSALTQPASV SGSPGQSITI SCSGSSSNIG NNAVNWYQQL PGKAPKLLIY YDDLLPSGVS DRFSGSKSGT SAFLAISGLQ SEDEADYYCA AWDDSLNGPV FGGGTKLTVL 2. Exemplary Anti-CD2 Antibodies
EVQLQQSGPE LQRPGASVKL SCKASGYIFT EYYMYWVKQR PKQGLELVGRIDPEDGSIDY VEKFKKKATL TADTSSNTAY MQLSSLTSED TATYFCARGKFNYRFAYWGQ GTLVTVSS DVVLTQTPPT LLATIGQSVS ISCRSSQSLL HSSGNTYLNW LLQRTGQSPQ PLIYLVSKLE SGVPNRFSGS GSGTDFTLKI SGVEAEDLGV YYCMQFTHYPYTFGAGTKLE LK 3. Exemplary Anti-CD8 Antibodies
QVQLLESGPE LLKPGASVKM SCKASGYTFT DYNMHWVKQS HGKSLEWIGYIYPYTGGTGY NQKFKNKATL TVDSSSSTAY MELRSLTSED SAVYYCARNF RYTYWYFDVW GQGTTVTVSS DIVMTQSPAS LAVSLGQRAT ISCRASESVD SYDNSLMHWY QQKPGQPPKV LIYLASNLES GVPARFSGSG SRTDFTLTID PVEADDAATY YCQQNNEDPYTFGGGTKLEI KR QVQLVESGGG VVQPGRSLRL SCAASGFTFS DFGMNWVRQA PGKGLEWVALIYYDGSNKFY ADSVKGRFTI SRDNSKNTLY LQMNSLRAED TAVYYCAKPHYDGYYHFFDS WGQGTLVTVS S DIQMTQSPSS LSASVGDRVT ITCKGSQDIN NYLAWYQQKP GKAPKLLIYNTDILHTGVPS RFSGSGSGTD FTFTISSLQP EDIATYYCYQ YNNGYTFGQG TKVEIK VI. Exemplary Disease Antigens
A. Exemplary Cancer Antigens Arrayed on the Surface of Cancer Cells
Antibody and Antibody-Based Molecules Antibody Name Cancer Antigens Therapeutic Target Application 3F8 Gd2 Neuroblastoma 8H9 B7-H3 Neuroblastoma, Sarcoma, Metastatic Brain Cancers Abagovomab CA-125 Ovarian Cancer Adecatumumab Epcam Prostate and Breast Cancer Afutuzumab CD20 Lymphoma Alacizumab VEGFR2 Cancer Altumomab CEA Colorectal Cancer Amatuximab Mesothelin Cancer Anatumomab TAG-72 Non-Small Cell Lung Carcinoma Mafenatox Anifrolumab Interferon A/B Systemic Lupus Erythematosus Receptor Anrukinzumab IL-13 Cancer Apolizumab HLA-DR Hematological Cancers Arcitumomab CEA Gastrointestinal Cancer Atinumab RTN4 Cancer Bectumomab CD22 Non-Hodgkin's Lymphoma (Detection) Belimumab BAFF Non-Hodgkin Lymphoma Bevacizumab VEGF-A Metastatic Cancer, Retinopathy of Prematurity Bivatuzumab CD44 V6 Squamous Cell Carcinoma Blinatumomab CD19 Cancer Brentuximab CD30 (TNFRSF8) Hematologic Cancers Cantuzumab MUC1 Cancers Cantuzumab Mucin Canag Colorectal Cancer Mertansine Caplacizumab VWF Cancers Capromab Prostatic Prostate Cancer (Detection) Carcinoma Cells Carlumab MCP-1 Oncology/Immune Indications Catumaxomab Epcam, CD3 Ovarian Cancer, Malignant Ascites, Gastric Cancer Cc49 Tag-72 Tumor Detection Cetuximab EGFR Metastatic Colorectal Cancer and Head and Neck Cancer Ch.14.18 Undetermined Neuroblastoma Citatuzumab Epcam Ovarian Cancer and other Solid Tumors Cixutumumab IGF-1 Receptor Solid Tumors Clivatuzumab MUC1 Pancreatic Cancer Conatumumab TRAIL-R2 Cancer Dacetuzumab CD40 Hematologic Cancers Dalotuzumab Insulin-Like Cancer Growth Factor I Receptor Daratumumab CD38 Cancer Demcizumab DLL4 Cancer Detumomab B-Lymphoma Cell Lymphoma Drozitumab DR5 Cancer Duligotumab HER3 Cancer Dusigitumab ILGF2 Cancer Ecromeximab GD3 Ganglioside Malignant Melanoma Eculizumab C5 Paroxysmal Nocturnal Hemoglobinuria Edrecolomab Epcam Colorectal Carcinoma Elotuzumab SLAMF7 Multiple Myeloma Elsilimomab IL-6 Cancer Enavatuzumab TWEAK Receptor Cancer Enlimomab ICAM-1 (CD54) Cancer Enokizumab IL9 Asthma Enoticumab DLL4 Cancer Ensituximab 5AC Cancer Epitumomab Episialin Cancer Cituxetan Epratuzumab CD22 Cancer, SLE Ertumaxomab HER2/neu, CD3 Breast Cancer Etaracizumab Integrin Avβ3 Melanoma, Prostate Cancer, Ovarian Cancer Faralimomab Interferon Receptor Cancer Farletuzumab Folate Receptor 1 Ovarian Cancer Fasinumab HNGF Cancer Fbta05 CD20 Chronic Lymphocytic Leukaemia Ficlatuzumab HGF Cancer Figitumumab IGF-1 Receptor Adrenocortical Carcinoma, Non-Small Cell Lung Carcinoma Flanvotumab TYRP1 Melanoma (Glycoprotein 75) Fontolizumab IFN-γ Crohn's Disease Fresolimumab TGF-B Idiopathic Pulmonary Fibrosis, Focal Segmental Glomerulosclerosis, Cancer Futuximab EGFR Cancer Galiximab CD80 B Cell Lymphoma Ganitumab IGF-I Cancer Gemtuzumab CD33 Acute Myelogenous Leukemia Ozogamicin Gevokizumab IL-β Diabetes Girentuximab Carbonic Clear Cell Renal Cell Carcinoma Anhydrase 9 (CA-IX) Glembatumumab GPNMB Melanoma, Breast Cancer Vedotin Golimumab TNF-A Rheumatoid Arthritis, Psoriatic Arthritis, Ankylosing Spondylitis Ibritumomab CD20 Non-Hodgkin's Lymphoma Tiuxetan Icrucumab VEGFR-1 Cancer Igovomab CA-125 Ovarian Cancer (Diagnosis) Imab362 Cldn18.2 Gastrointestinal Adenocarcinomas and Pancreatic Tumor Imgatuzumab EGFR Cancer Inclacumab Selectin P Cancer Indatuximab SDC1 Cancer Ravtansine Inotuzumab CD22 Cancer Ozogamicin Intetumumab CD51 Solid Tumors (Prostate Cancer, Melanoma) Ipilimumab CD152 Melanoma Iratumumab CD30 (TNFRSF8) Hodgkin's Lymphoma Itolizumab CD6 Cancer Labetuzumab CEA Colorectal Cancer Lambrolizumab PDCD1 Antineoplastic Agent Lampalizumab CFD Cancer Lexatumumab TRAIL-R2 Cancer Libivirumab Hepatitis B Surface Hepatitis B Antigen Ligelizumab IGHE Cancer Lintuzumab CD33 Cancer Lirilumab KIR2D Cancer Lorvotuzumab CD56 Cancer Lucatumumab CD40 Multiple Myeloma, Non-Hodgkin's Lymphoma, Hodgkin's Lymphoma Lumiliximab CD23 Chronic Lymphocytic Leukemia Mapatumumab TRAIL-R1 Cancer Margetuximab Ch4d5 Cancer Matuzumab EGFR Colorectal, Lung and Stomach Cancer Milatuzumab CD74 Multiple Myeloma and Other Hematological Malignancies Minretumomab TAG-72 Cancer Mitumomab GD3 Ganglioside Small Cell Lung Carcinoma Mogamulizumab CCR4 Cancer Morolimumab Rhesus Factor Cancer Moxetumomab CD22 Cancer Pasudotox Nacolomab C242 Antigen Colorectal Cancer Tafenatox Namilumab C SF2 Cancer Naptumomab 5T4 Non-Small Cell Lung Carcinoma, Estafenatox Renal Cell Carcinoma Narnatumab RON Cancer Nebacumab Endotoxin Sepsis Necitumumab EGFR Non-Small Cell Lung Carcinoma Nerelimomab TNF-A Cancer Nesvacumab Angiopoietin 2 Cancer Nimotuzumab EGFR Squamous Cell Carcinoma, Head and Neck Cancer, Nasopharyngeal Cancer, Glioma Nivolumab PD-1 Cancer Nofetumomab Undetermined Cancer Merpentan Ocaratuzumab CD20 Cancer Ofatumumab CD20 Chronic Lymphocytic Leukemia Olaratumab PDGF-R A Cancer Olokizumab IL6 Cancer Onartuzumab Human Scatter Cancer Factor Receptor Kinase Ontuxizumab TEM1 Cancer Oportuzumab Epcam Cancer Monatox Oregovomab CA-125 Ovarian Cancer Orticumab Oxldl Cancer Otlertuzumab CD37 Cancer Panitumumab EGFR Colorectal Cancer Pankomab Tumor Specific Ovarian Cancer Glycosylation of MUC1 Parsatuzumab EGFL7 Cancer Patritumab HER3 Cancer Pembrolizumab PD-1 Cancer Pemtumomab MUC1 Cancer Perakizumab IL17A Arthritis Pertuzumab HER2/neu Cancer Pidilizumab PD-1 Cancer and Infectious Diseases Pinatuzumab CD22 Cancer Vedotin Pintumomab Adenocarcinoma Adenocarcinoma Antigen Placulumab Human TNF Cancer Polatuzumab CD79B Cancer Vedotin Pritoxaximab Cancer Toxin Type-1 Pritumumab Vimentin Brain Cancer Quilizumab IGHE Cancer Racotumomab N- Cancer Glycolylneuraminic Acid Radretumab Fibronectin Extra Cancer Domain-B Ramucirumab VEGFR2 Solid Tumors Rilotumumab HGF Solid Tumors Rituximab CD20 Lymphomas, Leukemias, Some Autoimmune Disorders Robatumumab IGF-1 Receptor Cancer Roledumab RHD Cancer Samalizumab CD200 Cancer Satumomab TAG-72 Cancer Pendetide Seribantumab ERBB3 Cancer Setoxaximab Cancer Toxin Type-1 Sgn-CD19a CD19 Acute Lymphoblastic Leukemia and B Cell Non-Hodgkin Lymphoma Sgn-CD33 a CD33 Acute Myeloid Leukemia Sibrotuzumab FAP Cancer Siltuximab IL-6 Cancer Solitomab Epcam Cancer Sontuzumab Episialin Cancer Tab alumab BAFF B Cell Cancers Tacatuzumab Alpha-Fetoprotein Cancer Tetraxetan Taplitumomab CD19 Cancer Paptox Telimomab Undetermined Cancer Tenatumomab Tenascin C Cancer Teneliximab CD40 Cancer Teprotumumab CD221 Hematologic Tumors Ticilimumab CTLA-4 Cancer Tigatuzumab TRAIL-R2 Cancer Tnx-650 I1-13 Hodgkin's Lymphoma Tositumomab CD20 Follicular Lymphoma Tovetumab CD140a Cancer Trastuzumab HER2/neu Breast Cancer Trbs07 Gd2 Melanoma Tremelimumab CTLA-4 Cancer Tucotuzumab Epcam Cancer Celmoleukin Ublituximab MS4A1 Cancer Urelumab 4-1BB Cancer Vantictumab Frizzled Receptor Cancer Vapaliximab AOC3 (VAP-1) Cancer Vatelizumab ITGA2 Cancer Veltuzumab CD20 Non-Hodgkin's Lymphoma Vesencumab NRP1 Cancer Volociximab Integrin A5β1 Solid Tumors Vorsetuzumab CD70 Cancer Votumumab Tumor Antigen Colorectal Tumors CTAA16.88 Zalutumumab EGFR Squamous Cell Carcinoma of The Head And Neck Zatuximab HER1 Cancer Ziralimumab CD147 Cancer 1. Exemplary Anti-B7-H3 Antibodies
QVQLVQSGAE VKKPGASVKV SCKASGYTFT SYWMQWVRQA PGQGLEWMGTIYPGDGDTRY TQKFKGRVTI TADKSTSTAY MELSSLRSED TAVYYCARRGIPRLWYFDVW GQGTTVTVSS QVQLVQSGAE VKKPGASVKV SCKASGYTFT SYWMQWVRQA PGQGLEWMGTIYPGGGDTRY TQKFQGRVTI TADKSTSTAY MELSSLRSED TAVYYCARRGIPRLWYFDVW GQGTTVTVSS DIQMTQSPSS LSASVGDRVT ITCRASQDIS NYLNWYQQKP GKAPKLLIYYTSRLHSGVPS RFSGSGSGTD FTLTISSLQP EDIATYYCQQ GNTLPPTFGG GTKLEIK DIQMTQSPSS LSASVGDRVT ITCRASQSIS SYLNWYQQKP GKAPKLLIYYTSRLQSGVPS RFSGSGSGTD FTLTISSLQP EDIATYYCQQ GNTLPPTFGG GTKLEIK EVQLVESGGG LVKPGGSLRL SCAASGFTFS SYGMSWVRQA PGKGLEWVATINSGGSNTYY PDSLKGRFTI SRDNAKNSLY LQMNSLRAED TAVYYCARHDGGAMDYWGQG TTVTVSS DIQMTQSPSS LSASVGDRVT ITCRASESIY SYLAWYQQKP GKAPKLLVYNTKTLPEGVPS RFSGSGSGTD FTLTISSLQP EDFATYYCQH HYGTPPWTFG QGTRLEIK EVQLVESGGG LVQPGGSLRL SCAASGFTFS SFGMHWVRQA PGKGLEWVAYISSGSGTIYY ADTVKGRFTI SRDNAKNSLY LQMNSLRAED TAVYYCARHGYRYEGFDYWG QGTTVTVSS DIQMTQSPSF LSASVGDRVT ITCKASQNVD TNVAWYQQKP GKAPKALIYSASYRYSGVPS RFSGSGSGTD FTLTISSLQP EDFAEYFCQQ YNNYPFTFGQ GTKLEIK EVQLVESGGG LVQPGGSLRL SCAASGFTFS SFGMHWVRQA PGKGLEWVAYISSDSSAIYY ADTVKGRFTI SRDNAKNSLY LQMNSLRDED TAVYYCGRGRENIYYGSRLD YWGQGTTVTV SS DIQLTQSPSF LSASVGDRVT ITCKASQNVD TNVAWYQQKP GKAPKALIYSASYRYSGVPS RFSGSGSGTD FTLTISSLQP EDFATYYCQQ YNNYPFTFGQ GTKLEIK 2. Exemplary Anti-CEACAM5 and Anti-CEACAM6 Antibodies
QVQLQQSGPE VVRPGVSVKI SCKGSGYTFT DYAMHWVKQS HAKSLEWIGLISTYSGDTKY NQNFKGKATM TVDKSASTAY MELSSLRSED TAVYYCARGDYSGSRYWFAY WGQGTLVTVS S DIQMTQSPSS LSASVGDRVT ITCGASENIY GALNWYQRKP GKSPKLLIWGASNLADGMPS RFSGSGSGRQ YTLTISSLQP EDVATYYCQN VLSSPYTFGG GTKLEIK QVQLVESGGG VVQPGRSLRL SCSSSGFALT DYYMSWVRQA PGKGLEWLGFIANKANGHTT DYSPSVKGRF TISRDNSKNT LFLQMDSLRP EDTGVYFCAR DMGIRWNFDV WGQGTPVTVS S DIQLTQSPSS LSASVGDRVT MTCSASSRVS YIHWYQQKPG KAPKRWIYGTSTLASGVPAR FSGSGSGTDF TFTISSLQPE DIATYYCQQW SYNPPTFGQG TKVEIKR 3. Exemplary Anti-EGRF Antibodies
QVQLKQSGPG LVQPSQSLSI TCTVSGFSLT NYGVHWVRQS PGKGLEWLGVIWSGGNTDYN TPFTSRLSIN KDNSKSQVFF KMNSLQSNDT AIYYCARALTYYDYEFAYWG QGTLVTVSA DILLTQSPVI LSVSPGERVS FSCRASQSIG TNIHWYQQRT NGSPRLLIKYASESISGIPS RFSGSGSGTD FTLSINSVES EDIADYYCQQ NNNWPTTFGA GTKLELKR QVQLQESGPG LVKPSETLSL TCTVSGGSVSSGDYYWTWIR QSPGKGLEWI GHIYYSGNTNYNPSLKSRLT ISIDTSKTQF SLKLSSVTAA DTAIYYCVRDRVTGAFDIWG QGTMVTVSS DIQMTQSPSS LSASVGDRVT ITCQASQDISNYLNWYQQKP GKAPKLLIYDASNLETGVPS RFSGSGSGTD FTFTISSLQP EDIATYFCQHFDHLPLAFGG GTKVEIKR 4. Exemplary Anti-EphA2 Antibodies
QVQLKESGPG LVAPSQSLSI TCTVSGFSLS RYSVHWVRQP PGKGLEWLGMIWGGGSTDYNSALKSRLSIS KDNSKSQVFL KMNSLQTDDT AMYYCARKHGNYYTMDYWGQ GTSVTVSS DIQMTQTTSS LSASLGDRIT ISCRASQDIS NYLNWYQQKP DGTVKLLIYYTSRLHSGVPS RFSGSGSGTD YSLTISNLEQ EDIATYFCQQ GYTLYTFGGG TKLEIK QIQLVQSGPE LKKPGETVKI SCKASGFTFT NYGMNWVKQA PGKGLKWMGWINTYIGEPTY ADDFKGRFVF SLETSASTAY LQINNLKNED MATYFCARELGPYYFDYWGQ GTTLTVSS DVVMTQTPLS LPVSLGDQAS ISCRSSQSLV HSSGNTYLHW YLQKPGQSPK LLIYKVSNRF SGVPDRFSGS GSGTDFTLKI SRVEAEDLGV YFCSQSTHVPTFGSGTKLEI K EVQLVESGGG SVKPGGSLKL SCAASGFTFT DHYMYWVRQT PEKRLEWVATISDGGSFTSY PDSVKGRFTI SRDIAKNNLY LQMSSLKSED TAMYYCTRDESDRPFPYWGQ GTLVTVSS DIVLTQSHRS MSTSVGDRVN ITCKASQDVT TAVAWYQQKP GQSPKLLIFWASTRHAGVPD RFTGSGSGTD FTLTISSVQA GDLALYYCQQ HYSTPYTFGG GTKLEIK 5. Exemplary Anti-gpA33 Antibodies
QVQLVQSGAE VKKPGASVKV SCKASGYTFT GSWMNWVRQA PGQGLEWIGRIYPGDGETNY NGKFKDRVTI TADKSTSTAY MELSSLRSED TAVYYCARIYGNNVYFDVWG QGTTVTVSS DIQLTQSPSF LSASVGDRVT ITCSARSSIS FMYWYQQKPG KAPKLLIYDTSNLASGVPSR FSGSGSGTEF TLTISSLEAE DAATYYCQQW SSYPLTFGQG TKLEIK 6. Exemplary Anti HER2/Neu Antibodies
QVQLQQSGPE LVKPGASLKL SCTASGFNIK DTYIHWVKQR PEQGLEWIGRIYPTNGYTRY DPKFQDKATI TADTSSNTAY LQVSRLTSED TAVYYCSRWGGDGFYAMDYW GQGASVTVSS DIVMTQSHKF MSTSVGDRVS ITCKASQDVN TAVAWYQQKP GHSPKLLIYSASFRYTGVPD RFTGSRSGTD FTFTISSVQA EDLAVYYCQQ HYTTPPTFGG GTKVEIK EVQLVESGGG LVQPGGSLRL SCAASGFNIK DTYIHWVRQA PGKGLEWVARIYPTNGYTRY ADSVKGRFTI SADTSKNTAY LQMNSLRAED TAVYYCSRWGGDGFYAMDYW GQGTLVTVSS DIQMTQSPSS LSASVGDRVT ITCRASQDVN TAVAWYQQKP GKAPKLLIYSASFLYSGVPS RFSGSRSGTD FTLTISSLQP EDFATYYCQQ HYTTPPTFGQ GTKVEIK EVQLVESGGG LVQPGGSLRL SCAASGFTFT DYTMDWVRQA PGKGLEWVADVNPNSGGSIY NQRFKGRFTL SVDRSKNTLY LQMNSLRAED TAVYYCARNLGPSFYFDYWG QGTLVTVSS DIQMTQSPSS LSASVGDRVT ITCKASQDVS IGVAWYQQKP GKAPKLLIYSASYRYTGVPS RFSGSGSGTD FTLTISSLQP EDFATYYCQQ YYIYPYTFGQ GTKVEIK 7. Exemplary Anti-VEGF Antibodies
EVQLVESGGG LVQPGGSLRL SCAASGYTFT NYGMNWVRQA PGKGLEWVGWINTYTGEPTY AADFKRRFTF SLDTSKSTAY LQMNSLRAED TAVYYCAKYPHYYGSSHWYF DVWGQGTLVT VSS DIQMTQSPSS LSASVGDRVT ITCSASQDIS NYLNWYQQKP GKAPKVLIYFTSSLHSGVPS RFSGSGSGTD FTLTISSLQP EDFATYYCQQ YSTVPWTFGQ GTKVEIKR 8. Exemplary Anti-5T4 Antibodies
QVQLVQSGAE VKKPGASVKV SCKASGYTFTSFWMHWVRQA PGQGLEWMGRIDPNRGGTEYNEKAKSRVTM TADKSTSTAY MELSSLRSED TAVYYCAGGNPYYPMDYWGQ GTTVTVSS DIQMTQSPSS LSASVGDRVT ITCRASQGISNYLAWFQQKP GKAPKSLIYRANRLQSGVPS RFSGSGSGTD FTLTISSLQP EDVATYYCLQYDDFPWTFGQ GTKLEIK QVQLQQPGAE LVKPGASVKM SCKASGYTFTSYWITWVKQR PGQGLEWIGDIYPGSGRANYNEKFKSKATL TVDTSSSTAY MQLSSLTSED SAVYNCARYGPLFTTVVDPNSYAMDYWGQG TSVTVSS DVLMTQTPLS LPVSLGDQAS ISCRSSQSIVYSNGNTYLEW YLQKPGQSPK LLIYKVSNRFSGVPDRFSGS GSGTDFTLKI SRVEAEDLGV YYCFQGSHVPFTFGSGTKLE IK 9. Exemplary Anti-IL13Rα2 Antibodies
EVQLVESGGG LVQPGGSLRL SCAASGFTFS RNGMSWVRQA PGKGLEWVATVSSGGSYIYY ADSVKGRFTI SRDNAKNSLY LQMNSLRAED TAVYYCARQGTTALATRFFD VWGQGTLVTV SS DIQMTQSPSS LSASVGDRVT ITCKASQDVG TAVAWYQQKP GKAPKLLIYSASYRSTGVPS RFSGSGSGTD FTLTISSLQP EDFATYYCQH HYSAPWTFGG GTKVEIK 10. Exemplary Anti-CD123 Antibodies
EVQLVQSGAE LKKPGASVKV SCKASGYTFT DYYMKWVRQA PGQGLEWIGDIIPSNGATFYNQKFKGRVTI TVDKSTSTAY MELSSLRSED TAVYYCARSHLLRASWFAYW GQGTLVTVSS DFVMTQSPDS LAVSLGERVT MSCKSSQSLL NSGNQKNYLT WYQQKPGQPP KLLIYWASTR ESGVPDRFSG SGSGTDFTLT ISSLQAEDVA VYYCQNDYSYPYTFGQGTKL EIK 11. Exemplary Anti-CD19 Antibodies
QVTLRESGPA LVKPTQTLTL TCTFSGFSLS TSGMGVGWIR QPPGKALEWL AHIWWDDDKRYNPALKSRLT ISKDTSKNQV FLTMTNMDPV DTATYYCARMELWSYYFDYW GQGTTVTVSS ENVLTQSPAT LSVTPGEKAT ITCRASQSVS YMHWYQQKPG QAPRLLIYDASNRASGVPSR FSGSGSGTDH TLTISSLEAE DAATYYCFQG SVYPFTFGQG TKLEIK B. Exemplary Pathogen-Associated Antigens
1. Exemplary Anti-HIV Antibodies
QVQLVQSGGG VFKPGGSLRL SCEASGFTFT EYYMTWVRQA PGKGLEWLAY ISKNGEYSKYSPSSNGRFTI SRDNAKNSVF LQLDRLSADD TAVYYCARADGLTYFSELLQYIFDLWGQGA RVTVSS DIVMTQSPDS LAVSPGERAT IHCKSSQTLLYSSNNRHSIA WYQQRPGQPP KLLLYWASMRLSGVPDRFSG SGSGTDFTLT INNLQAEDVA IYYCHQYSSHPPTFGHGTRV EIK QVQLQESGPG LVKPSQTLSL SCTVSGGSSS SGAHYWSWIR QYPGKGLEWI GYIHYSGNTYYNPSLKSRIT ISQHTSENQF SLKLNSVTVA DTAVYYCARGTRLRTLRNAF DIWGQGTLVT VSS QSALTQPPSA SGSPGQSVTI SCTGTSSDVG GYNYVSWYQH HPGKAPKLII SEVNNRPSGV PDRFSGSKSG NTASLTVSGL QAEDEAEYYC SSYTDIHNFV FGGGTKLTVL 2. Exemplary Anti-RSV Antibody
QVTLRESGPA LVKPTQTLTL TCTFSGFSLS TSGMSVGWIR QPPGKALEWL ADIWWDDKKDYNPSLKSRLT ISKDTSKNQV VLKVTNMDPA DTATYYCARSMITNWYFDVW GAGTTVTVSS DIQMTQSPST LSASVGDRVT ITCRASQSVGYMHWYQQKPG KAPKLLIYDTSKLASGVPSR FSGSGSGTEF TLTISSLQPD DFATYYCFQGSGYPFTFGGG TKLEIK VII. Exemplary Binding Molecules of the Present Invention
DART-A 1 151 Trastuzumab VL 125 CD16-M1 VH 64 2 152 CD16-M1 VL 65 Trastuzumab VH 124 3 153 Common Diabody Polypeptide Chain DART-B 1 154 Trastuzumab VL 125 CD16-M2 VH 75 2 155 CD16-M2 VL 76 Trastuzumab VH 124 3 153 Common Diabody Polypeptide Chain DART-C 1 156 Trastuzumab VL 125 hCD16-M1 VH 72 2 157 hCD16-M1 VL 73 Trastuzumab VH 124 3 153 Common Diabody Polypeptide Chain DART-D 1 158 Trastuzumab VL 125 hCD16-M2 VH1 83 2 159 hCD16-M2 VL 85 Trastuzumab VH 124 3 153 Common Diabody Polypeptide Chain DART-E 1 160 Trastuzumab VL 125 hCD16-M2 VH2 84 2 159 hCD16-M2 VL 85 Trastuzumab VH 124 3 153 Common Diabody Polypeptide Chain DART-F 1 161 A32 VL 143 hCD16-M1 VH 72 2 162 hCD16-M1 VL 73 A32 VH 142 3 153 Common Diabody Polypeptide Chain DART-G 1 163 A32 VL 143 hCD16-M2 VH1 83 2 164 hCD16-M2 VL 85 A32 VH 142 3 153 Common Diabody Polypeptide Chain DART-H 1 165 7B2 VL 141 hCD16-M1 VH 72 2 166 hCD16-M1 VL 73 7B2 VH 140 3 153 Common Diabody Polypeptide Chain DART-I 1 186 Trastuzumab VL 125 hCD16-M1A VH 58 2 157 hCD16-M1 VL 73 Trastuzumab VH 124 3 153 Common Diabody Polypeptide Chain DART-J 1 156 Trastuzumab VL 125 hCD16-M1 VH 72 2 187 hCD16-M1B VL 59 Trastuzumab VH 124 3 153 Common Diabody Polypeptide Chain DART-K 1 186 Trastuzumab VL 125 hCD16-M1A VH 58 2 187 hCD16-M1B VL 59 Trastuzumab VH 124 3 153 Common Diabody Polypeptide Chain DART-L 1 188 CD19 mAb 1 VL 139 hCD16-M1 VH 72 2 189 hCD16-M1 VL 73 CD19 mAb 1 VH 138 3 153 Common Diabody Polypeptide Chain DART-M 1 188 CD19 mAb 1 VL 139 hCD16-M1 VH 72 2 190 hCD16-M1B VL 59 CD19 mAb 1 VH 138 3 153 Common Diabody Polypeptide Chain DART-N 1 191 CD19 mAb 1 VL 139 hCD16-M1A VH 58 2 190 hCD16-M1B VL 59 CD19 mAb 1 VH 138 3 153 Common Diabody Polypeptide Chain DART-1 1 167 Trastuzumab VL 125 h3G8 VH 62 2 168 h3G8 VL 63 Trastuzumab VH 124 3 153 Common Diabody Polypeptide Chain DART-2 1 — vPalivizumab VL 145 h3G8 VH 62 2 — h3G8 VL 63 vPalivizumab VH 144 3 153 Common Diabody Polypeptide Chain DART-3 1 169 vPalivizumab VL 145 hCD16-M1 VH 72 2 170 hCD16-M1 VL 73 vPalivizumab VH 144 3 153 Common Diabody Polypeptide Chain DART-4 1 — 7B2 VL 141 h3G8 VH 62 2 — h3G8 VL 63 7B2 VH 140 3 153 Common Diabody Polypeptide Chain DART-5 1 — vPalivizumab VL 145 hCD16-M1 VH 72 2 — hCD16-M1 VL 73 vPalivizumab VH 144 3 153 Common Diabody Polypeptide Chain DART-6 1 — vPalivizumab VL 145 hCD16-M1 VH 72 2 — hCD16-M1B VL 59 vPalivizumab VH 144 3 153 Common Diabody Polypeptide Chain DART-7 1 — vPalivizumab VL 145 hCD16-M1A VH 58 2 — hCD16-M1B VL 59 vPalivizumab VH 144 3 153 Common Diabody Polypeptide Chain DART-X 1 171 7B2 VL 141 CD16-M1 VH 64 2 172 CD16-M1 VL 65 7B2 VH 140 DART-Y 1 173 7B2 VL 141 CD16-M2 VH 75 2 174 CD16-M2 VL 76 7B2 VH 140 DART-Z 1 — A32 VL 143 4-LSN1 VH 175 2 — 4-LSN1 VL 176 A32 VH 142 DART-0 1 — 7B2 VL 141 h3G8 VH 62 2 — h3G8 VL 63 7B2 VH 140 A. CD16 x HER2/neu Binding Molecule, DART-A
DIQMTQSPSS LSASVGDRVT ITCRASQDVN TAVAWYQQKP GKAPKLLIYS ASFLYSGVPS RFSGSRSGTD FTLTISSLQP EDFATYYCQQ HYTTPPTFGQ GTKVEIKGGGSGGGGEVKLV ESGGTLVKPG GSLKLSCAAS GFTFNNYGMS WVRQTPEKRL EWVATISGGG SYTFYPDSVK GRFTISRDNA KNSLYLQMSS LRSEDTALYY CIRQSARAPE PYWGQGTLVT VSSASTKGEV AACEKEVAAL EKEVAALEKE VAALEKGGGD KTHTCPPCPA PEAAGGPSVF LFPPKPKDTL MISRTPEVTC VVVDVSHEDP EVKFNWYVDG VEVHNAKTKP REEQYNSTYR VVSVLTVLHQ DWLNGKEYKC KVSNKALPAP IEKTISKAKG QPREPQVYTL PPSREEMTKN QVSLWCLVKG FYPSDIAVEW ESNGQPENNY KTTPPVLDSD GSFFLYSKLT VDKSRWQQGN VFSCSVMHEA LHNHYTQKSL SLSPGK DIVMTQSQKF MSTSVGDRVS VTCKASQNVG THVAWYQQKS GQSPKSLLYS ASYRYSGVPD RFSGSGSGTD FTLTISNVQS EDLAEYFCQQ YKSYPLTFGA GTKLELKGGGSGGGGEVQLV ESGGGLVQPG GSLRLSCAAS GFNIKDTYIH WVRQAPGKGL EWVARIYPTN GYTRYADSVK GRFTISADTS KNTAYLQMNS LRAEDTAVYY CSRWGGDGFY AMDYWGQGTL VTVSSASTKG KVAACKEKVA ALKEKVAALK EKVAALKE DKTHTCPPCP APEAAGGPSV FLFPPKPKDT LMISRTPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT LPPSREEMTK NQVSLSCAVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLVSKL TVDKSRWQQG NVFSCSVMHE ALHNRYTQKS LSLSPGK B. CD16 x HER2/neu Binding Molecule, “DART-B”
DIQMTQSPSS LSASVGDRVT ITCRASQDVN TAVAWYQQKP GKAPKLLIYS ASFLYSGVPS RFSGSRSGTD FTLTISSLQP EDFATYYCQQ HYTTPPTFGQ GTKVEIK EVQLQ QSGPELVKPG ASVKMSCKAS GYTFTSSAMH WVKKNPGQGL EWIGYINHYN DGIKYNERFK GKATLTSDKS SSTAYMELSS LTSEDSAVYY CATGYRYASW FASWGQGTLV TVSSASTKGE VAACEKEVAA LEKEVAALEK EVAALEKGGG DKTHTCPPCP APEAAGGPSV FLFPPKPKDT LMISRTPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT LPPSREEMTK NQVSLWCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE ALHNHYTQKS LSLSPGK DILLTQSPAI LSVSPGERVS FSCRASQNIG TSIHWYQQRT DGSPRLLIKS VSESISGIPS RFSGSGSGTD FTLTINGVES GDISDYYCQQ SNSWPLTFGA GTKLELK EVQLV ESGGGLVQPG GSLRLSCAAS GFNIKDTYIH WVRQAPGKGL EWVARIYPTN GYTRYADSVK GRFTISADTS KNTAYLQMNS LRAEDTAVYY CSRWGGDGFY AMDYWGQGTL VTVSSASTKG KVAACKEKVA ALKEKVAALK EKVAALKE C. CD16 x HER2/neu Binding Molecule “DART-C”
DIQMTQSPSS LSASVGDRVT ITCRASQDVN TAVAWYQQKP GKAPKLLIYS ASFLYSGVPS RFSGSRSGTD FTLTISSLQP EDFATYYCQQ HYTTPPTFGQ GTKVEIK EVQLV ESGGGLVKPG GSLRLSCAAS GFTFSNYGMS WVRQAPGKGL EWVATISGGG SYTFYPDSVK GRFTISRDNA KNSLYLQMNS LRTEDTALYY CVRQSARAPE PYWGQGTLVT VSSASTKGEV AACEKEVAAL EKEVAALEKE VAALEKGGGD KTHTCPPCPA PEAAGGPSVF LFPPKPKDTL MISRTPEVTC VVVDVSHEDP EVKFNWYVDG VEVHNAKTKP REEQYNSTYR VVSVLTVLHQ DWLNGKEYKC KVSNKALPAP IEKTISKAKG QPREPQVYTL PPSREEMTKN QVSLWCLVKG FYPSDIAVEW ESNGQPENNY KTTPPVLDSD GSFFLYSKLT VDKSRWQQGN VFSCSVMHEA LHNHYTQKSL SLSPGK DIQMTQSPSF LSASVGDRVT ITCRASQNVG THVAWYQQKP GKAPKSLLYS ASYRYSGVPS RFSGSGSGTD FTLTISSLQS EDIATYYCQQ YKSYPLTFGQ GTKLEIK EVQLV ESGGGLVQPG GSLRLSCAAS GFNIKDTYIH WVRQAPGKGL EWVARIYPTN GYTRYADSVK GRFTISADTS KNTAYLQMNS LRAEDTAVYY CSRWGGDGFY AMDYWGQGTL VTVSSASTKG KVAACKEKVA ALKEKVAALK EKVAALKE D. CD16 x HER2/neu Binding Molecule “DART-D”
DIQMTQSPSS LSASVGDRVT ITCRASQDVN TAVAWYQQKP GKAPKLLIYS ASFLYSGVPS RFSGSRSGTD FTLTISSLQP EDFATYYCQQ HYTTPPTFGQ GTKVEIK QVQLV QSGAEVKKPG ASVKVSCKAS GYTFTSSAMH WVRQAPGQGL EWMGYINHYN DGIKYNERFK GRVTITADKS TSTAYMELSS LRSEDTAVYY CATGYRYASW FASWGQGTLV TVSSASTKGE VAACEKEVAA LEKEVAALEK EVAALEKGGG DKTHTCPPCP APEAAGGPSV FLFPPKPKDT LMISRTPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT LPPSREEMTK NQVSLWCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE ALHNHYTQKS LSLSPGK EIVLTQSPAT LSVSPGERAT LSCRASQNIG TSIHWYQQKP DQSPKLLIKS VSESISGVPS RFSGSGSGTD FTLTINSLEA EDFATYYCQQ SNSWPLTFGQ GTKLEIK EVQLV ESGGGLVQPG GSLRLSCAAS GFNIKDTYIH WVRQAPGKGL EWVARIYPTN GYTRYADSVK GRFTISADTS KNTAYLQMNS LRAEDTAVYY CSRWGGDGFY AMDYWGQGTL VTVSSASTKG KVAACKEKVA ALKEKVAALK EKVAALKE E. CD16 x HER2/Neu Binding Molecule “DART-E”
DIQMTQSPSS LSASVGDRVT ITCRASQDVN TAVAWYQQKP GKAPKLLIYS ASFLYSGVPS RFSGSRSGTD FTLTISSLQP EDFATYYCQQ HYTTPPTFGQ GTKVEIK QVQLV QSGAEVKKPG ASVKVSCKAS GYTFTSSAMH WVRQAPGQGL EWMGYINHYN DGIKYNERFK GRVTITADKS TSTAYMELSS LRSEDTAVYY CARGYRYASW FASWGQGTLV TVSSASTKGE VAACEKEVAA LEKEVAALEK EVAALEKGGG DKTHTCPPCP APEAAGGPSV FLFPPKPKDT LMISRTPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT LPPSREEMTK NQVSLWCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE ALHNHYTQKS LSLSPGK F. CD16 x HIV Env Binding Molecule “DART-F”
QSALTQPPSA SGSPGQSVTI SCTGTSSDVG GYNYVSWYQH HPGKAPKLII SEVNNRPSGV PDRFSGSKSG NTASLTVSGL QAEDEAEYYC SSYTDIHNFV FGGGTKLTVL EV QLVESGGGLV KPGGSLRLSC AASGFTFSNY GMSWVRQAPG KGLEWVATIS GGGSYTFYPD SVKGRFTISR DNAKNSLYLQ MNSLRTEDTA LYYCVRQSAR APEPYWGQGT LVTVSSASTKGEVAACEKEV AALEKEVAAL EKEVAALEKG GGDKTHTCPP CPAPEAAGGP SVFLFPPKPK DTLMISRTPE VTCVVVDVSH EDPEVKFNWY VDGVEVHNAK TKPREEQYNS TYRVVSVLTV LHQDWLNGKE YKCKVSNKAL PAPIEKTISK AKGQPREPQV YTLPPSREEM TKNQVSLWCL VKGFYPSDIA VEWESNGQPE NNYKTTPPVL DSDGSFFLYS KLTVDKSRWQ QGNVFSCSVM HEALHNHYTQ KSLSLSPGK DIQMTQSPSF LSASVGDRVT ITCRASQNVG THVAWYQQKP GKAPKSLLYS ASYRYSGVPS RFSGSGSGTD FTLTISSLQS EDIATYYCQQ YKSYPLTFGQ GTKLEIK VQLQ ESGPGLVKPS QTLSLSCTVS GGSSSSGAHY WSWIRQYPGK GLEWIGYIHY SGNTYYNPSL KSRITISQHT SENQFSLKLN SVTVADTAVY YCARGTRLRT LRNAFDIWGQ GTLVTVSSASTKGKVAACKE KVAALKEKVA ALKEKVAALK E G. CD16 x HIV Env Binding Molecule “DART-G”
QSALTQPPSA SGSPGQSVTI SCTGTSSDVG GYNYVSWYQH HPGKAPKLII SEVNNRPSGV PDRFSGSKSG NTASLTVSGL QAEDEAEYYC SSYTDIHNFV FGGGTKLTVL QV QLVQSGAEVK KPGASVKVSC KASGYTFTSS AMHWVRQAPG QGLEWMGYIN HYNDGIKYNE RFKGRVTITA DKSTSTAYME LSSLRSEDTA VYYCATGYRY ASWFASWGQG TLVTVSSASTKGEVAACEKE VAALEKEVAA LEKEVAALEK GGGDKTHTCP PCPAPEAAGG PSVFLFPPKP KDTLMISRTP EVTCVVVDVS HEDPEVKFNW YVDGVEVHNA KTKPREEQYN STYRVVSVLT VLHQDWLNGK EYKCKVSNKA LPAPIEKTIS KAKGQPREPQ VYTLPPSREE MTKNQVSLWC LVKGFYPSDI AVEWESNGQP ENNYKTTPPV LDSDGSFFLY SKLTVDKSRW QQGNVFSCSV MHEALHNHYT QKSLSLSPGK EIVLTQSPAT LSVSPGERAT LSCRASQNIG TSIHWYQQKP DQSPKLLIKS VSESISGVPS RFSGSGSGTD FTLTINSLEA EDFATYYCQQ SNSWPLTFGQ GTKLEIK QVQLQ ESGPGLVKPS QTLSLSCTVS GGSSSSGAHY WSWIRQYPGK GLEWIGYIHY SGNTYYNPSL KSRITISQHT SENQFSLKLN SVTVADTAVY YCARGTRLRT LRNAFDIWGQ GTLVTVSSASTKGKVAACKE KVAALKEKVA ALKEKVAALK E H. CD16 x HIV Env Binding Molecule “DART-H”
DIVMTQSPDS LAVSPGERAT IHCKSSQTLL YSSNNRHSIA WYQQRPGQPP KLLLYWASMR LSGVPDRFSG SGSGTDFTLT INNLQAEDVA IYYCHQYSSH PPTFGHGTRV EIK EVQLVESGG GLVKPGGSLR LSCAASGFTF SNYGMSWVRQ APGKGLEWVA TISGGGSYTF YPDSVKGRFT ISRDNAKNSL YLQMNSLRTE DTALYYCVRQ SARAPEPYWG QGTLVTVSSASTKGEVAACE KEVAALEKEV AALEKEVAAL EKGGGDKTHT CPPCPAPEAA GGPSVFLFPP KPKDTLMISR TPEVTCVVVD VSHEDPEVKF NWYVDGVEVH NAKTKPREEQ YNSTYRVVSV LTVLHQDWLN GKEYKCKVSN KALPAPIEKT ISKAKGQPRE PQVYTLPPSR EEMTKNQVSL WCLVKGFYPS DIAVEWESNG QPENNYKTTP PVLDSDGSFF LYSKLTVDKS RWQQGNVFSC SVMHEALHNH YTQKSLSLSP GK DIQMTQSPSF LSASVGDRVT ITCRASQNVG THVAWYQQKP GKAPKSLLYS ASYRYSGVPS RFSGSGSGTD FTLTISSLQS EDIATYYCQQ YKSYPLTFGQ GTKLEIK QVQLV QSGGGVFKPG GSLRLSCEAS GFTFTEYYMT WVRQAPGKGL EWLAYISKNG EYSKYSPSSN GRFTISRDNA KNSVFLQLDR LSADDTAVYY CARADGLTYF SELLQYIFDL WGQGARVTVS SASTKGKVAA CKEKVAALKE KVAALKEKVA ALKE I. CD16 x HER2/Neu Binding Molecule “DART-I”
DIQMTQSPSS LSASVGDRVT ITCRASQDVN TAVAWYQQKP GKAPKLLIYS ASFLYSGVPS RFSGSRSGTD FTLTISSLQP EDFATYYCQQ HYTTPPTFGQ GTKVEIK EVQLV ESGGGLVKPG GSLRLSCAAS GFTFSNYGMS WVRQAPGKGL EWVATISGGG SYTFYPDSVK GRFTISRDNA KNSLYLQMNS LRTEDTALYY CVRQSANSPV PYWGQGTLVT VSS EV AACEKEVAAL EKEVAALEKE VAALEKGGGD KTHTCPPCPA PEAAGGPSVF LFPPKPKDTL MISRTPEVTC VVVDVSHEDP EVKFNWYVDG VEVHNAKTKP REEQYNSTYR VVSVLTVLHQ DWLNGKEYKC KVSNKALPAP IEKTISKAKG QPREPQVYTL PPSREEMTKN QVSLWCLVKG FYPSDIAVEW ESNGQPENNY KTTPPVLDSD GSFFLYSKLT VDKSRWQQGN VFSCSVMHEA LHNHYTQKSL SLSPGK J. CD16 x HER2/Neu Binding Molecule “DART-J”
DIQMTQSPSF LSASVGDRVT ITCRASQNVG THVAWYQQKP GKAPKSLLYS ASYRYSGVPS RFSGSGSGTD FTLTISSLQS EDIATYYCQD YTNYPLTFGQ GTKLEIK EVQLV ESGGGLVQPG GSLRLSCAAS GFNIKDTYIH WVRQAPGKGL EWVARIYPTN GYTRYADSVK GRFTISADTS KNTAYLQMNS LRAEDTAVYY CSRWGGDGFY AMDYWGQGTL VTVSSASTKG KVAACKEKVA ALKEKVAALK EKVAALKE K. CD16 x HER2/Neu Binding Molecule “DART-K”
L. CD16 x CD19 Binding Molecule “DART-L”
ENVLTQSPAT LSVTPGEKAT ITCRASQSVS YMHWYQQKPG QAPRLLIYDA SNRASGVPSR FSGSGSGTDH TLTISSLEAE DAATYYCFQG SVYPFTFGQG TKLEIK EVQLVE SGGGLVKPGG SLRLSCAASG FTFSNYGMSW VRQAPGKGLE WVATISGGGS YTFYPDSVKG RFTISRDNAK NSLYLQMNSL RTEDTALYYC VRQSARAPEP YWGQGTLVTV SS EV AACEKEVAAL EKEVAALEKE VAALEKGGGDKTHTCPPCPA PEAAGGPSVF LFPPKPKDTL MISRTPEVTC VVVDVSHEDP EVKFNWYVDG VEVHNAKTKP REEQYNSTYR VVSVLTVLHQ DWLNGKEYKC KVSNKALPAP IEKTISKAKG QPREPQVYTL PPSREEMTKN QVSLWCLVKG FYPSDIAVEW ESNGQPENNY KTTPPVLDSD GSFFLYSKLT VDKSRWQQGN VFSCSVMHEA LHNHYTQKSL SLSPGK DIQMTQSPSF LSASVGDRVT ITCRASQNVG THVAWYQQKP GKAPKSLLYS ASYRYSGVPS RFSGSGSGTD FTLTISSLQS EDIATYYCQQ YKSYPLTFGQ GTKLEIK QVTLR ESGPALVKPT QTLTLTCTFS GFSLSTSGMG VGWIRQPPGK ALEWLAHIWW DDDKRYNPAL KSRLTISKDT SKNQVFLTMT NMDPVDTATY YCARMELWSY YFDYWGQGTT VTVSSGGCGG GKVAACKEKV AALKEKVAAL KEKVAALKE M. CD16 x CD19 Binding Molecule “DART-M”
DIQMTQSPSF LSASVGDRVT ITCRASQNVG THVAWYQQKP GKAPKSLLYS ASYRYSGVPS RFSGSGSGTD FTLTISSLQS EDIATYYCQD YTNYPLTFGQ GTKLEIK QVTLR ESGPALVKPT QTLTLTCTFS GFSLSTSGMG VGWIRQPPGK ALEWLAHIWW DDDKRYNPAL KSRLTISKDT SKNQVFLTMT NMDPVDTATY YCARMELWSY YFDYWGQGTT VTVSSGGCGGGKVAACKEKV AALKEKVAAL KEKVAALKE N. CD16 x CD19 Binding Molecule “DART-N”
ENVLTQSPAT LSVTPGEKAT ITCRASQSVS YMHWYQQKPG QAPRLLIYDA SNRASGVPSR FSGSGSGTDH TLTISSLEAE DAATYYCFQG SVYPFTFGQG TKLEIK EVQLVE SGGGLVKPGG SLRLSCAASG FTFSNYGMSW VRQAPGKGLE WVATISGGGS YTFYPDSVKG RFTISRDNAK NSLYLQMNSL RTEDTALYYC VRQSANSPVP YWGQGTLVTV SSGGCGGGEV AACEKEVAAL EKEVAALEKE VAALEKGGGD KTHTCPPCPA PEAAGGPSVF LFPPKPKDTL MISRTPEVTC VVVDVSHEDP EVKFNWYVDG VEVHNAKTKP REEQYNSTYR VVSVLTVLHQ DWLNGKEYKC KVSNKALPAP IEKTISKAKG QPREPQVYTL PPSREEMTKN QVSLWCLVKG FYPSDIAVEW ESNGQPENNY KTTPPVLDSD GSFFLYSKLT VDKSRWQQGN VFSCSVMHEA LHNHYTQKSL SLSPGK O. CD16 x HER2/Neu Binding Molecule “DART-1”
QVTLRESGPA LVKPTQTLTL TCTFSGFSLS TSGMGVGWIR QPPGKALEWL AHIWWDDDKRYNPALKSRLT ISKDTSKNQV VLTMTNMDPV DTATYYCAQI NPAWFAYWGQ GTLVTVSS DIVMTQSPDS LAVSLGERAT INCKASQSVDFDGDSFMNWY QQKPGQPPKL LIYTTSNLES GVPDRFSGSG SGTDFTLTIS SLQAEDVAVY YCQQSNEDPY TFGQGTKLEI K
instead of the VH and VL Domains of anti-human CD16 antibody CD16-M1 (SEQ ID NO:64 and SEQ ID NO:65, respectively) that are present in the DART-A. The VH and VL Domains of h3G8 are used herein as a comparator CD16 binding site. DART-1 is composed of three polypeptide chains having two Binding Domains that comprises the VL and VH Domains of anti-human CD16 antibody CD16-M1 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of Trastuzumab (and is thus immunospecific for an epitope of the HER2/neu Cancer Antigen). The three polypeptide chains associate to form a covalently bonded DART® diabody capable of immunospecifically binding the epitope of CD16 and the epitope of the HER2/neu Cancer Antigen (see, e.g., DIQMTQSPSS LSASVGDRVT ITCRASQDVN TAVAWYQQKP GKAPKLLIYS ASFLYSGVPS RFSGSRSGTD FTLTISSLQP EDFATYYCQQ HYTTPPTFGQ GTKVEIK QVTLR ESGPALVKPT QTLTLTCTFS GFSLSTSGMG VGWIRQPPGK ALEWLAHIWW DDDKRYNPAL KSRLTISKDT SKNQVVLTMT NMDPVDTATY YCAQINPAWF AYWGQGTLVT VSSASTKGEV AACEKEVAAL EKEVAALEKE VAALEKGGGD KTHTCPPCPA PEAAGGPSVF LFPPKPKDTL MISRTPEVTC VVVDVSHEDP EVKFNWYVDG VEVHNAKTKP REEQYNSTYR VVSVLTVLHQ DWLNGKEYKC KVSNKALPAP IEKTISKAKG QPREPQVYTL PPSREEMTKN QVSLWCLVKG FYPSDIAVEW ESNGQPENNY KTTPPVLDSD GSFFLYSKLT VDKSRWQQGN VFSCSVMHEA LHNHYTQKSL SLSPGK DIVMTQSPDS LAVSLGERAT INCKASQSVD FDGDSFMNWY QQKPGQPPKL LIYTTSNLES GVPDRFSGSG SGTDFTLTIS SLQAEDVAVY YCQQSNEDPY TFGQGTKLEI K E VQLVESGGGL VQPGGSLRLS CAASGFNIKD TYIHWVRQAP GKGLEWVARI YPTNGYTRYA DSVKGRFTIS ADTSKNTAYL QMNSLRAEDT AVYYCSRWGG DGFYAMDYWG QGTLVTVSSA STKGKVAACK EKVAALKEKV AALKEKVAAL KE P. CD16 x RSV Binding Molecule “DART-2”
Q. CD16 x RSV Binding Molecule “DART-3”
DIQMTQSPST LSASVGDRVT ITCRASQSVG YMHWYQQKPG KAPKLLIYDT SKLASGVPSR FSGSGSGTEF TLTISSLQPD DFATYYCFQG SGYPFTFGGG TKLEIK EVQLVE SGGGLVKPGG SLRLSCAASG FTFSNYGMSW VRQAPGKGLE WVATISGGGS YTFYPDSVKG RFTISRDNAK NSLYLQMNSL RTEDTALYYC VRQSARAPEP YWGQGTLVTV SSASTKGEVA ACEKEVAALE KEVAALEKEV AALEKGGGDK THTCPPCPAP EAAGGPSVFL FPPKPKDTLM ISRTPEVTCV VVDVSHEDPE VKFNWYVDGV EVHNAKTKPR EEQYNSTYRV VSVLTVLHQD WLNGKEYKCK VSNKALPAPI EKTISKAKGQ PREPQVYTLP PSREEMTKNQ VSLWCLVKGF YPSDIAVEWE SNGQPENNYK TTPPVLDSDG SFFLYSKLTV DKSRWQQGNV FSCSVMHEAL HNHYTQKSLS LSPGK DIQMTQSPSF LSASVGDRVT ITCRASQNVG THVAWYQQKP GKAPKSLLYS ASYRYSGVPS RFSGSGSGTD FTLTISSLQS EDIATYYCQQ YKSYPLTFGQ GTKLEIK QVTLR ESGPALVKPT QTLTLTCTFS GFSLSTSGMS VGWIRQPPGK ALEWLADIWW DDKKDYNPSL KSRLTISKDT SKNQVVLKVT NMDPADTATY YCARSMITNW YFDVWGAGTT VTVSSASTKG KVAACKEKVA ALKEKVAALK EKVAALKE R. CD16 x HIV Env Binding Molecule “DART-4
S. CD16 x RSV Binding Molecule “DART-5”
T. CD16 x RSV Binding Molecule “DART-6”
U. CD16 x RSV Binding Molecule “DART-7”
V. CD16 x HIV Env Molecule “DART-X”
DIVMTQSPDS LAVSPGERAT IHCKSSQTLL YSSNNRHSIA WYQQRPGQPP KLLLYWASMR LSGVPDRFSG SGSGTDFTLT INNLQAEDVA IYYCHQYSSH PPTFGHGTRV EIK EVKLVESGG TLVKPGGSLK LSCAASGFTF NNYGMSWVRQ TPEKRLEWVA TISGGGSYTF YPDSVKGRFT ISRDNAKNSL YLQMSSLRSE DTALYYCIRQ SARAPEPYWG QGTLVTVSSA STKGEVAACE KEVAALEKEV AALEKEVAAL EK DIVMTQSQKF MSTSVGDRVS VTCKASQNVG THVAWYQQKS GQSPKSLLYS ASYRYSGVPD RFSGSGSGTD FTLTISNVQS EDLAEYFCQQ YKSYPLTFGA GTKLELK QVQLV QSGGGVFKPG GSLRLSCEAS GFTFTEYYMT WVRQAPGKGL EWLAYISKNG EYSKYSPSSN GRFTISRDNA KNSVFLQLDR LSADDTAVYY CARADGLTYF SELLQYIFDL WGQGARVTVS SASTKGKVAA CKEKVAALKE KVAALKEKVA ALKE W. CD16 x HIV Env Binding Molecule “DART-Y”
DIVMTQSPDS LAVSPGERAT IHCKSSQTLL YSSNNRHSIA WYQQRPGQPP KLLLYWASMR LSGVPDRFSG SGSGTDFTLT INNLQAEDVA IYYCHQYSSH PPTFGHGTRV EIK EVQLQQSGP ELVKPGASVK MSCKASGYTF TSSAMHWVKK NPGQGLEWIG YINHYNDGIK YNERFKGKAT LTSDKSSSTA YMELSSLTSE DSAVYYCATG YRYASWFASW GQGTLVTVSS ASTKGEVAAC EKEVAALEKE VAALEKEVAA LEK DILLTQSPAI LSVSPGERVS FSCRASQNIG TSIHWYQQRT DGSPRLLIKS VSESISGIPS RFSGSGSGTD FTLTINGVES GDISDYYCQQ SNSWPLTFGA GTKLELK QVQLV QSGGGVFKPG GSLRLSCEAS GFTFTEYYMT WVRQAPGKGL EWLAYISKNG EYSKYSPSSN GRFTISRDNA KNSVFLQLDR LSADDTAVYY CARADGLTYF SELLQYIFDL WGQGARVTVS SASTKGKVAA CKEKVAALKE KVAALKEKVA ALKE X. CD16 x HIV Env Binding Molecule “DART-0”
Y. CD16 x HIV Env Binding Molecule “DART-Z”
EEVQLVQSGA EVKKPGESLK VSCKASGYTF TSYYMHWVRQ APGQGLEWMG IINPSGGSTS YAQKFQGRVT MTRDTSTSTV YMELSSLRSE DTAVYYCARG SAYYYDFADY WGQGTLVTVS S
VL Domain of Anti-Human CD16A scFv 4-LS21 (SEQ ID NO:176):
QPVLTQPSSV SVAPGQTATI SCGGHNIGSK NVHWYQQRPG QSPVLVIYQD NKRPSGIPER FSGSNSGNTA TLTISGTQAM DEADYYCQVW DNYSVLFGGG TKLTVL
instead of the VH and VL Domains of anti-human CD16 antibody CD16-M1 (SEQ ID NO:64 and SEQ ID NO:65, respectively) that are present in DART-X. Thus, DART-Z is composed of two polypeptide chains having one Binding Domain that comprises the VL and VH Domains of the anti-human CD16A scFv 4-LS21 (and is thus immunospecific for an epitope of CD16) and one Binding Domain that comprises the VL and VH Domains of A32 (and is thus immunospecific for an epitope of the HIV Env protein). These polypeptide chains associate to form a covalently bonded DART® diabody composed of two polypeptide chains that possesses one Binding Domain immunospecific for the epitope of CD16 and one Binding Domain immunospecific for the epitope of the HIV env protein (see, e.g., Z. CD16 x HIV Env x HIV Env Trivalent Molecules
TRIDENT-A 1 177 A32 VL 143 hCD16-M1 VH 72 2 178 hCD16-M1 VL 73 A32 VH 142 3 179 A32 VH 142 4 180 A32 VL 143 TRIDENT-B 1 177 A32 VL 143 hCD16-M1 VH 72 2 178 hCD16-M1 VL 73 A32 VH 142 3 181 7B2 VH 140 4 182 7B2 VL 141 1. CD16 x HIV Env x HIV Env Trivalent Molecule “TRIDENT-A”
QSALTQPPSA SGSPGQSVTI SCTGTSSDVG GYNYVSWYQH HPGKAPKLII SEVNNRPSGV PDRFSGSKSG NTASLTVSGL QAEDEAEYYC SSYTDIHNFV FGGGTKLTVL EV QLVESGGGLV KPGGSLRLSC AASGFTFSNY GMSWVRQAPG KGLEWVATIS GGGSYTFYPD SVKGRFTISR DNAKNSLYLQ MNSLRTEDTA LYYCVRQSAR APEPYWGQGT LVTVSSASTK GEVAACEKEV AALEKEVAAL EKEVAALEKG GGDKTHTCPP CPAPEAAGGP SVFLFPPKPK DTLMISRTPE VTCVVVDVSH EDPEVKFNWY VDGVEVHNAK TKPREEQYNS TYRVVSVLTV LHQDWLNGKE YKCKVSNKAL PAPIEKTISK AKGQPREPQV YTLPPSREEM TKNQVSLWCL VKGFYPSDIA VEWESNGQPE NNYKTTPPVL DSDGSFFLYS KLTVDKSRWQ QGNVFSCSVM HEALHNHYTQ KSLSLSPGK DIQMTQSPSF LSASVGDRVT ITCRASQNVG THVAWYQQKP GKAPKSLLYS ASYRYSGVPS RFSGSGSGTD FTLTISSLQS EDIATYYCQQ YKSYPLTFGQ GTKLEIK QVQLQ ESGPGLVKPS QTLSLSCTVS GGSSSSGAHY WSWIRQYPGK GLEWIGYIHY SGNTYYNPSL KSRITISQHT SENQFSLKLN SVTVADTAVY YCARGTRLRT LRNAFDIWGQ GTLVTVSSAS TKGKVAACKE KVAALKEKVA ALKEKVAALK E QVQLQESGPG LVKPSQTLSL SCTVSGGSSS SGAHYWSWIR QYPGKGLEWI GYIHYSGNTY YNPSLKSRIT ISQHTSENQF SLKLNSVTVA DTAVYYCARG TRLRTLRNAF DIWGQGTLVT VSSASTKGPS VFPLAPSSKS TSGGTAALGC LVKDYFPEPV TVSWNSGALT SGVHTFPAVL QSSGLYSLSS VVTVPSSSLG TQTYICNVNH KPSNTKVDKR VEPKSCDKTHTCPPCPAPEA AGGPSVFLFP PKPKDTLMIS RTPEVTCVVV DVSHEDPEVK FNWYVDGVEV HNAKTKPREE QYNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKALPAPIEK TISKAKGQPR EPQVYTLPPS REEMTKNQVS LSCAVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF FLVSKLTVDK SRWQQGNVFS CSVMHEALHN RYTQKSLSLS PGK QSALTQPPSA SGSPGQSVTI SCTGTSSDVG GYNYVSWYQH HPGKAPKLII SEVNNRPSGV PDRFSGSKSG NTASLTVSGL QAEDEAEYYC SSYTDIHNFV FGGGTKLTVL RTVAAPSVFI FPPSDEQLKS GTASVVCLLN NFYPREAKVQ WKVDNALQSG NSQESVTEQD SKDSTYSLSS TLTLSKADYE KHKVYACEVT HQGLSSPVTK SFNRGEC 2. CD16 x HIV Env x HIV Env Trivalent Molecule “TRIDENT-B”
QVQLVQSGGG VFKPGGSLRL SCEASGFTFT EYYMTWVRQA PGKGLEWLAY ISKNGEYSKY SPSSNGRFTI SRDNAKNSVF LQLDRLSADD TAVYYCARAD GLTYFSELLQ YIFDLWGQGA RVTVSSASTK GPSVFPLAPS SKSTSGGTAA LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP AVLQSSGLYS LSSVVTVPSS SLGTQTYICN VNHKPSNTKV DKRVEPKSCDKTHTCPPCPA PEAAGGPSVF LFPPKPKDTL MISRTPEVTC VVVDVSHEDP EVKFNWYVDG VEVHNAKTKP REEQYNSTYR VVSVLTVLHQ DWLNGKEYKC KVSNKALPAP IEKTISKAKG QPREPQVYTL PPSREEMTKN QVSLSCAVKG FYPSDIAVEW ESNGQPENNY KTTPPVLDSD GSFFLVSKLT VDKSRWQQGN VFSCSVMHEA LHNRYTQKSL SLSPGK DIVMTQSPDS LAVSPGERAT IHCKSSQTLL YSSNNRHSIA WYQQRPGQPP KLLLYWASMR LSGVPDRFSG SGSGTDFTLT INNLQAEDVA IYYCHQYSSH PPTFGHGTRV EIKRTVAAPS VFIFPPSDEQ LKSGTASVVC LLNNFYPREA KVQWKVDNAL QSGNSQESVT EQDSKDSTYS LSSTLTLSKA DYEKHKVYAC EVTHQGLSSP VTKSFNRGEC VIII. Methods of Production
IX. Uses of the Binding Molecules of the Present Invention
X. Pharmaceutical Compositions
XI. Methods of Administration
EXAMPLES
Example 1
Characterization of CD16 Binding Molecules
DART-A CD16 × HER2/neu CD16-M1 Trastuzumab DART-B CD16 × HER2/neu CD16-M2 Trastuzumab DART-C CD16 × HER2/neu hCD16-M1 Trastuzumab DART-D CD16 × HER2/neu hCD16-M2 (VH1) Trastuzumab DART-E CD16 × HER2/neu hCD16-M2 (VH2) Trastuzumab DART-F CD16 × HIV hCD16-M1 A32 DART-G CD16 × HIV hCD16-M2 (VH1) A32 DART-H CD16 × HIV hCD16-M1 7B2 DART-I CD16 × HER2/neu hCD16-M1A Trastuzumab DART-J CD16 × HER2/neu hCD16-M1B Trastuzumab DART-K CD16 × HER2/neu hCD16-M1AB Trastuzumab DART-L CD16 × CD19 hCD16-M1 CD19 mAb 1 DART-M CD16 × CD19 hCD16-M1B CD19 mAb 1 DART-N CD16 × CD19 hCD16-M1AB CD19 mAb 1 DART-1 CD16 × HER2/neu h3G8 Trastuzumab DART-2 CD16 × RSV h3G8 Palivizumab variant DART-3 CD16 × RSV hCD16-M1 vPalivizumab DART-4 CD16 × HIV h3G8 7B2 DART-5 CD16 × RSV hCD16-M1 vPalivizumab DART-6 CD16 × RSV hCD16-M1B vPalivizumab DART-7 CD16 × RSV hCD16-M1AB vPalivizumab DART-X CD16 × HIV CD16-M1 7B2 DART-Y CD16 × HIV CD16-M2 7B2 DART-Z CD16 × HIV 4-LSN1 A32 DART-0 CD16 × HIV h3G8 7B2 Human CD16A 158F DART-1 1.7 × 106 1.1 × 10−1 65.9 DART-C 1.6 × 106 3.0 × 10−4 0.2 DART-D 4.7 × 105 1.2 × 10−3 2.5 DART-F 1.5 × 106 3.4 × 10−4 0.2 DART-G 4.7 × 105 1.2 × 10−3 2.6 Human CD16A 158V DART-1 1.3 × 106 9.0 × 10−3 6.9 DART-C 1.4 × 106 3.9 × 10−4 0.3 DART-D 5.2 × 105 1.2 × 10−3 2.3 DART-F 1.4 × 106 3.7 × 10−4 0.3 DART-G 5.4 × 105 1.2 × 10−3 2.2 Molecule ka kd KD(nM) Human CD16A V158F DART-A 4.2 × 106 3.3 × 10−4 0.1 DART-B 2.9 × 105 4.8 × 10−4 1.7 DART-1 5.3 × 105 3.4 × 10−2 63.1 DART-C 2.8 × 106 2.6 × 10−4 0.1 DART-D 4.3 × 105 1.0 × 10−3 2.4 DART-E 2.3 × 105 1.6 × 10−3 7.2 DART-F 4.2 × 106 2.4 × 10−4 0.1 DART-G 4.7 × 105 8.9 × 10−4 1.9 DART-H 4.5 × 106 1.9 × 10−4 0.04 DART-4 6.1 × 105 4.7× 10−2 76.8 DART-Z 2.9 × 105 4.3 × 10−3 15.2 Human CD16A 158V DART-A 3.9 × 106 4.5 × 10−4 0.1 DART-B 3.2 × 105 4.1 × 10−4 1.3 DART-1 2.4 × 105 6.5 × 10−3 26.7 DART-C 2.4 × 106 3.2 × 10−4 0.1 DART-D 5.6 × 105 7.1 × 10−4 1.3 DART-E 3.3 × 105 1.1 × 10−3 3.3 DART-F 4.0 × 106 4.0 × 10−4 0.1 DART-G 6.6 × 105 5.5 × 10−4 0.8 DART-H 3.9 × 106 3.7 × 10−4 0.1 DART-4 2.9 × 106 6.9 × 10−3 23.3 DART-Z 2.6 × 105 4.2 × 10−3 15.7 Human CD16B DART-A 2.6 × 106 1.3 × 10−3 0.5 DART-B 2.2 × 105 7.4 × 10−4 3.3 DART-1 8.9 × 105 5.5 × 10−3 6.1 DART-C 1.5 × 106 6.9 × 10−4 0.5 DART-D 3.5 × 105 1.3 × 10−3 3.6 DART-E 1.5 × 105 1.9 × 10−3 12.5 DART-F 2.8 × 106 1.1 × 10−3 0.4 DART-G 4.6 × 105 1.2 × 10−3 2.6 DART-H 2.5 × 106 8.9 × 10−4 0.4 DART-4 7.8 × 105 6.1 × 10−3 7.8 DART-Z N/A* N/A* N/A* * The CD16 Binding Domain of DART-Z derives from scFv 4-LS21, which is specific for CD16A Example 2
Binding of DART-F and DART-G CD16 Binding Molecules to NK Cells, Neutrophils and T Cells
Example 3
CD16B Allotype Specificity of Anti-CD16 Binding Molecules CD16-M1 and CD16-M2
Example 4
CD16B Allotype Specificity of CD16 Binding Molecules CD16-M1 and CD16-M2
Example 5
Binding of CD16 Binding Molecules to Human CD16, Cynomolgus Monkey CD16 and Murine CD16
DART-1 h3G8 13 29 No Binding DART-C hCD16-M1 12 571 No Binding DART-D hCD16-M2 4 597 No Binding Example 6
Evaluation of CD16 Binding Molecule-Mediated Cytotoxicity of HER2/Neu-Positive Cells
or a HER2/Neux RSV diabody (negative control) were separately incubated with target cancer cells expressing different levels of HER2/neu in the presence of Effector cells (either human PBMC (E:T=30:1) or purified human NK cells (E:T=2:1)) for 24 hours. The percentage cytotoxicity (i.e., redirected cell killing) was determined by measuring the release of lactate dehydrogenase (LDH) into the media by damaged cells essentially as described below. The results of this investigation are shown in Example 7
Evaluation of CD16 Binding Molecule-Mediated Cytotoxicity of HIV-Infected Cells
or DART-2 (a h3G8 x RSV diabody used as a negative control here)), were separately incubated with target 293HEK D371, which express HIV Env, in the presence of Effector cells (either human PBMC (E:T=30:1) or purified human NK cells (E:T=3:1) for 24 hours. The percentage cytotoxicity (i.e. redirected cell killing) was determined by measuring the release of lactate dehydrogenase (LDH) into the media by damaged cells using an LDH redirected cell killing assay essentially as described below. The results of this investigation are shown in
or DART-3 (a CD16 x RSV diabody having an hCD16-M1 CD16 Binding Domain used as a negative control here), were separately incubated with target HEK/D371 cells, which express the HIV env protein, in the presence of Effector cells (either Jurkat/CD16A 158F (Example 8
Optimization of Binding to Non-Human Primate CD16
24 hour Assay/LDH 48 hour Assay/LDH EC50, ng/mL EC50, ng/mL HuPBMCs CynoPBMCs HuPBMCs CynoPBMCs DART-1 131 170 156.1 101 DART-C 1.42 51.47 0.822 70.6 DART-I 1.05 36.98 0.492 13.31 DART-J 4.4 39.91 1.06 10.88 DART-K 10.8 29.44 2.24 6.66 Example 9
CD16 x CD19 Binding Molecules
Example 10
Exemplary Redirected Cell Killing Assays