Copper Complex for Capturing Carbon Dioxide
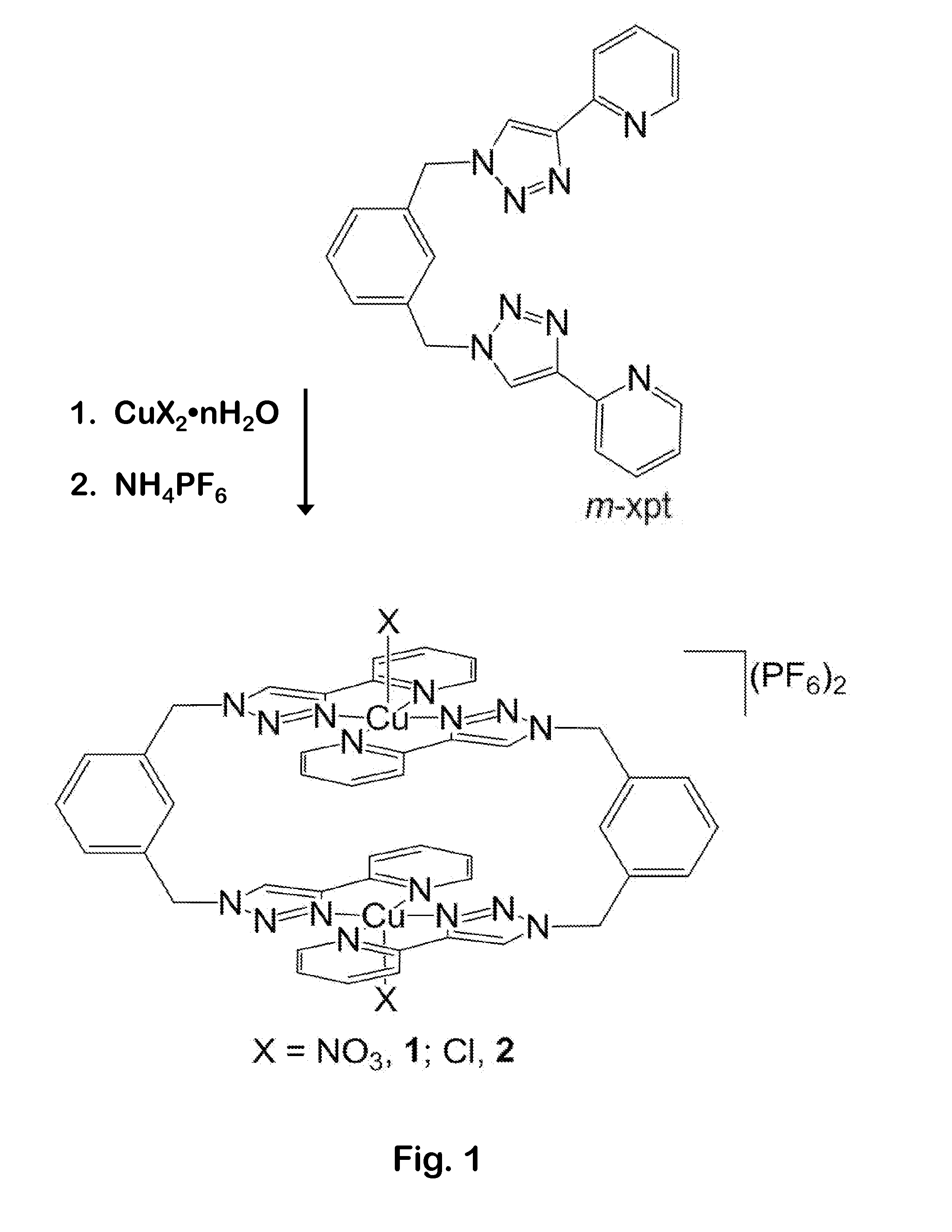
The benefit of the Oct. 14, 2013 filing date of U.S. provisional patent application Ser. No. 61/890,403 is claimed under 35 U.S.C. §119(e). The complete disclosure of the priority application is hereby incorporated by reference in its entirety. This invention was made with Government support under Louisiana EPSCoR award number EPS-1003897 awarded by the National Science Foundation. The United States Government has certain rights in the invention. This invention pertains to a composition and method for reducing carbon dioxide to oxalate. An economical route for converting carbon dioxide to value-added organic compounds would be highly desirable because of the role carbon dioxide (CO2) plays in global climate change, and in the depletion of fossil fuel resources. Although CO2is an inexpensive, non-toxic, abundant carbon feedstock, it is difficult to economically reduce CO2to a more useful form because of its thermodynamic stability and kinetic inertness. For CO2reduction to be attractive on a large scale, the process needs to work under mild reaction conditions, and the process must be economical. CO2has traditionally been captured by absorption into a solution of an organic amine. This method is energy-intensive; it requires heating the solution to disperse the absorbed CO2for storage. If the absorbed CO2is simply driven off, then it must be stored somewhere (e.g. in an underground rock formation) to avoid release into the atmosphere. Other methods that have been tried include activating and reducing CO2by electrochemical and electrocatalytic means in the presence of various transition metals and alloys. There have been reports of using low-valent d-block and f-block metal complexes to reduce CO2to oxalate. Horn, B., Limberg, C., Herwig, C. & Braun, B. Nickel(I)-mediated transformations of carbon dioxide in closed synthetic cycles: reductive cleavage and coupling of CO2generating NiICO, NilICO3and NiIIC2O4NiII) entities. Angamuthu, R., Byers, P., Lutz, M., Spek, A. L. & Bouwman, E. Electrocatalytic CO2Conversion to Oxalate by a Copper Complex. Science 327, 313-315, doi:10.1126/science.1177981 (2010) reported a binuclear copper(I) complex that can reduce CO2to oxalate, forming a tetranuclear copper(II) oxalate complex. Oxalate was then released by electrolysis, using lithium perchlorate as the supporting electrolyte, to complete the electrocatalytic cycle. The complex contained an amino-acid-derived ligand that bound two Cu atoms. Two of the complexes reacted with four CO2molecules to form two oxalates: 2Cu2L+4CO2→Cu4L2(C2O4)2. Crowley, J. D.; Bandeen, P. H., A multicomponent CuAAC “click” approach to a library of hybrid polydentate 2-pyridyl-1,2,3-triazole ligands: new building blocks for the generation of metallosupramolecular architectures. Other methods to reduce CO2include electrochemical or photochemical processes. In the Bocarsly “liquid light” approach, CO2reacts with an electrochemically-reduced solution of a heterocyclic amine such as pyridine. In photochemical reduction, some or all of the energy needed for CO2reduction is supplied by light. Reductive dimerization of carbon dioxide to oxalate (C2O42−) converts an environmental pollutant into a more useful organic compound. There is an unfilled need for improved, economical methods to reduce CO2to oxalate. If a suitable chemistry could operate rapidly and cleanly enough, then it could be used to capture CO2from the atmosphere or from other chemical processes (e.g. combustion, cement manufacture). Oxalate and oxalic acid have many uses, including in extractive metallurgy, as mordants in dyeing processes, as bleaching agents, as miticides, and as reagents in various synthetic processes. In very large quantities, oxalate may also simply be used to sequester CO2in solid form, e.g. as CaC2O4. We have discovered a novel composition and method for capturing CO2. A pollutant/greenhouse gas can be converted into a valuable organic compound using a nontoxic reducing agent. CO2may be economically captured from the atmosphere, from products of combustion, or from byproducts of various chemical processes. The novel method reduces CO2in a three-step reaction cycle, in which a binuclear metal-organic compound converts CO2to oxalate in a redox cycle under mild conditions. In one embodiment, a novel copper(II) complex [Cu2L2X2]2+ (L=m-xylylene-bis(pyridyltriazole); X=NO3, Cl) is reduced to its Cu(I) counterpart. This reduction may be performed either electrochemically or with a mild reducing agent such as sodium ascorbate. The [Cu2L2X2]2+ complex selectively reacts with CO2to reduce it to oxalate, in an oxalate-bridged binuclear complex [Cu2L2(μ-C2O4)]2+. The bound oxalate ion may be released as oxalic acid by treatment with a strong acid such as HCl or HNO3, thus regenerating the “empty” [Cu2L2X2]2+ complexes and completing the cycle. The novel metal-organic system will reduce CO2to oxalate under mild reaction conditions. The binuclear Cu complex provides a unique environment that promotes binding and reduction of two CO2molecules to produce one oxalate. Although CO2is fixed relatively slowly, the Cu(I) dimer selectively reacts with CO2instead of O2, a distinct advantage for a simple-to-operate process. Oxalic acid is readily released from the product. The binuclear metallacyclic copper complex selectively captures CO2from air (or other streams) and reduces it to oxalate in an oxalate-bridged complex. This complex releases oxalic acid when treated with dilute mineral acid, regenerating the “empty” macrocycle and completing the cyclic process. The complex reacts both with pure CO2, and with CO2in concentrations typical of those in air. In an alternative embodiment, we have used vitamin C (sodium ascorbate) as a reducing agent to yield the copper(I) complex that may then be used to reduce CO2. Sodium ascorbate is a milder reagent and is easier to use than many other reducing agents. All commercially available reagents and solvents were purchased from Aldrich or Alfa Aesar, and were used without further purification.1H Nuclear Magnetic Resonance (NMR) spectra were recorded on a Bruker AV-400 MHz spectrometer. ESI mass spectra were measured on an Agilent 6210 instrument. FTIR spectra were recorded on a Bruker Tensor 27 spectrometer in ATR mode. M-H-W Laboratories (Phoenix, Ariz.) performed elemental analyses. UV-visible spectra were recorded on an Aviv 14DS spectrometer. The samples were prepared in sealed flasks, and spectra were recorded using cuvettes connected to the flasks. Cyclic voltammetry measurements were performed using a Princeton Applied Research Model 273A potentiostat/galvanostat with Power Suite 2.53 software. The measurements were carried out in 0.1 M Bu4NP F6in dimethylformamide (DMF), with a glassy carbon working electrode, Ag/AgCl reference electrode, and Pt wire counter electrode. The ligand m-xpt was synthesized following the procedure of Pokharel, U. R., Fronczek, F. R. & Maverick, A. W. Cyclic pyridyltriazole-Cu(II) dimers as supramolecular hosts. To a stirred solution of Cu(NO3)2.3H2O (0.612 g, 2.54 mmol) in acetonitrile (75 mL), m-xpt (1.00 g, 2.54 mmol) in chloroform (50 mL) was added dropwise. The reaction mixture was stirred at room temperature for 2 hours. The precipitate was collected by filtration, washed with acetonitrile and chloroform, and dried to yield a blue solid [Cu2(m-xpt)2](NO3)2](NO3)2(1.57 g, 1.35 mmol). This solid was dissolved in water (200 mL), and an aqueous solution of NH4PF6(1.32 g, 8.09 mmol) was added. The mixture was stirred for 10 min and filtered. The precipitate was collected, washed with water, and dried in air to give compound 1 (1.84 g, 84%) as a light-blue powder. ESI-MS: m/z 1349.078, [Cu2(m-xpt)2(PF6)3]+ (calcd 1349.082). Anal. Calcd for [Cu2(m-xpt)2(NO3)](PF6)3H2O: C 36.93, H 2.68, N 16.64. Found: C 36.88, H 2.92, N 15.40. Complexing the ligand m-xpt with Cu(NO3)2gave the dimeric macrocycle [Cu2(m-xpt)2(NO3)2](NO3)2. Although the distance between the two Cu centers in this compound was appropriate for small-molecule guests, the compound was insoluble in common organic solvents. To improve solubility and to widen the scope of host-guest chemistry, we replaced two of the nitrate anions with the more hydrophobic PF6−. Metathesis gave [Cu2(m-xpt)2(NO3)2](PF6)2, Compound 1. A stirred solution of complex 1 (200 mg, 0.14 mmol) in 20 mL of DMF was reduced to [Cu2(m-xpt)2](PF6)2, Compound 3, by adding sodium ascorbate (41 mg, 0.21 mmol) under nitrogen for ca. 1 hour. The amount of nitrogen seen in the elemental analysis was consistently lower than expected. We postulate that this observation may be due to the presence of species such as [Cu2(m-xpt)2](PF6)4in the product mixture. When the product was crystallized by vapor diffusion of diethyl ether into its solution in DMF with benzene, we isolated [Cu2(m-xpt)2(NO3)2](PF6)2, 1. Complexing m-xpt with CuCl2gave the dimeric macrocycle [Cu2(m-xpt)2Cl2]Cl2. This compound was also insoluble in common organic solvents, and we replaced the chloride anions with PF6−. Metathesis gave [Cu2(m-xpt)2Cl2](PF6)2, Compound 2. To a stirred solution of [Cu2(m-xpt)2Cl2]Cl2(2.00 g, 1.86 mmol) in water (300 mL), excess NH4PF6(1.82 g, 11.2 mmol) was added. The mixture was stirred for 10 minutes. The precipitate was collected by filtration, washed with water, and dried to give complex 2 (2.05 g, 86%) as a blue-green solid. A crystalline product was obtained from DMF by vapor diffusion of diethyl ether. ESI-MS: 1239.085 [Cu2(m-xpt)2(PF6)2Cl]+ (calcd 1239.087). Anal. Calcd for [Cu2(m-xpt)2Cl2](PF6)2.3DMF: C 42.55, H 3.84, N 17.79, Cl 4.74. Found: C 42.92, H 3.49, N 17.92, Cl 5.21. To a stirred solution of complex 1 (200 mg, 0.14 mmol) or complex 2 (178 mg, 0.14 mmol) in DMF (20 mL), sodium ascorbate (41 mg, 0.21 mmol) was added under N2. After 1 hour, the yellow solution was slowly diffused with diethyl ether under nitrogen for 2 days. The resulting solid precipitate was collected by filtration, washed with diethyl ether, and dried to give complex 3 (164 mg, 85% from complex 1; 156 mg, 81% from complex 2).1H NMR (DMSO-d6, 400 MHz): 5.79 (s, 8H, CH2), 7.43-7.49 (m, 12H, Ar), 8.12 (br, 8H, Ar), 8.43 (br, 4H, Ar), 9.23 (br, 4H, triazole).13C NMR (DMSO-d6, 125 MHz): 54.3, 122.1, 125.1, 125.9, 128.8, 129.3, 130.1, 136.0, 139.3, 146.0, 147.1, 149.5. ESI-MS: 1059.171 [Cu2(m-xpt)2(PF6)]+ (calcd 1059.154). Anal. Calcd for [Cu2(m-xpt)2](PF6)2: C 43.82, H 3.01, N 18.58. Found: C 43.58, H 3.16, N 17.92. As we had observed for compound 1, compound 3 also gave microanalyses that were low in nitrogen. The addition of a small amount of ether to the formula gave better agreement; however, there were no signals attributable to ether or to other impurities in the compound's NMR spectrum. We tested the selectivity of the reaction of complex 3 with CO2, by generating the Cu(I) dimer. The yellow solutions that resulted from the previous syntheses were exposed to air to react with atmospheric CO2, and also to allow slow evaporation of the DMF solvent. After 4 to 5 days, we isolated complex 4 in high yield (96% from complex 1; 69% from complex 2). We observed green octahedral crystals of [Cu2(m-xpt)2(μ-C2O4)](PF6)2, 4, synthesized starting from [Cu2(m-xpt)2(NO3)2](PF6)2, 1 (crystal size ca. 0.3 mm). Both green and blue-green crystals were formed when complex 2 was used as the starting material. This conversion demonstrated selective reaction of [Cu2(m-xpt)2]2+, 3, with CO2over O2. Both types of crystals were characterized by X-ray crystallography. The green crystals were found to be the desired oxalate-bridged dimer 4. The blue-green crystalline product was the starting Cu(II) dimer, [Cu2(m-xpt)2Cl2](PF6)2. Both were washed with acetonitrile, which dissolved the blue-green crystals. Dimer 4 was collected by filtration (194 mg, 96% from complex 1; 138 mg, 69% from complex 2). FTIR (cm−1): 1670(s), 1645 (s), 1610 (s), 1454 (s), 839 (vs), 785 (vs), 715 (vs). Anal. Calcd for [Cu2(m-xpt)2(μ-C2O4)(PF6)2].2DMF: C 43.37, H 3.50, N 17.51. Found: C 43.48, H 3.52, N 17.58. The Cu . . . Cu separation, 5.4213(7) Å and the distances within the bridging oxalate ligand (C—C 1.544(7) Å; C—O 1.248(3) Å), were similar to those that have been observed for other C2O42−-bridged copper complexes. The Cu(I) complex 3 derived from complex 1 was transferred into a sealed flask connected to a cuvette. The reaction mixture was purged with nitrogen using four alternating cycles of vacuum and nitrogen. The reaction mixture showed no changes in the UV-vis spectrum after 48 hours. The N2was then replaced with13CO2. The yellow solution progressively turned green over a period of 128 hours, as13CO2was added periodically to maintain the pressure at ca. 1 atm. After the reaction was complete, the solution was poured into a watch glass and left to crystallize by slow evaporation of DMF. The crystalline solid was washed with acetone. As a complementary reaction to that displacing bound oxalate from complex 4, we also tested the ability of empty macrocycles to accept oxalate ion as a guest. The reaction of complex 1 and 2 with tetrabutylammonium oxalate in acetonitrile yielded [Cu2(m-xpt)2(μ-C2O4)]2+, as confirmed by single crystal X-ray analysis. We prepared [Cu2(m-xpt)2(μ-C2O4)]2+ from Cu(II) and oxalate as follows: To a stirred solution of complex 1 (200 mg, 0.14 mmol) or complex 2 (178 mg, 0.14 mmol) in acetonitrile (20 mL), tetrabutylammonium oxalate (80 mg, 0.15 mmol) was added. The mixture was stirred for 30 minutes and then left to crystallize by slow evaporation of the solvent. Yellow-green crystals formed, and were then washed with acetonitrile and air-dried to give 4a (178 mg, 87% from complex 1; 147 mg, 72% from complex 2). X-ray analysis showed that these crystals were the acetonitrile solvate of [Cu2(m-xpt)2(μ-C2O4)](PF6)2, 4a (Cu . . . Cu=5.462(2) Å), which is isostructural with the DMF solvate 4. (See Table 2 for data and refinement parameters.) Bound oxalate was removed from [Cu2(m-xpt)2(μ-C2O4)](PF6)2, 4 to regenerate the “empty” Cu(II) macrocycle [Cu2(m-xpt)2]4+. Crystallization from DMF/H2O gave the “empty” host complexes [Cu2(m-xpt)2Cl2]Cl2and [Cu2(m-xpt)2(H2O)2](NO3)4, 5, from the reaction of complex 4 with HCl and HNO3, respectively. The yields of empty complexes after acid treatment were nearly quantitative: 94% (HCl) and 96%(HNO3). HCl(aq) (8 eq) or HNO3(aq) (8 eq) (2 mL of 0.32 M, 0.64 mmol) was added to a stirred suspension of complex 4 (100 mg, 0.07 mmol) in methanol (50 mL). The solid dissolved, giving a greenish-yellow solution (HCl) or a blue solution (HNO3), which deposited a green or blue-green precipitate after stirring for an additional 3 hours. The mixture was filtered and the solid was re-dissolved in a mixture of water and DMF (4:1 v/v). The solution was poured into a watch glass and left to stand for slow evaporation. After 4 to 5 days, a blue-green (HCl) or blue (HNO3) crystalline product had formed. These crystals were washed with ca. 5 mL methanol and were air-dried. X-ray analysis of the products revealed the formation of empty Cu(II) macrocycles: [Cu2(m-xpt)2Cl2]Cl2.4DMF (with HCl; 92 mg, 94%) or [Cu2(m-xpt)2(H2O)2](NO3)4.4DMF, 5 (with HNO3; 81 mg, 96%). The filtrate from the above reaction was evaporated to dryness. The solid was dissolved in water (2 mL). The solution was neutralized with KOH(aq) and extracted with chloroform. The aqueous phase was acidified with HCl and again evaporated to dryness to give oxalic acid as a white solid which was characterized by13C NMR (163.2 ppm) and FTIR spectroscopy (νCO=1668 cm−1; with13CO2, 1642 cm−1). [Cu2(m-xpt)2(H2O)2](NO3)4, 5, formed by removal of oxalate from [Cu2(m-xpt)2(μ-C2O4)](PF6)2, 4 had a Cu . . . Cu distance=7.2441(8) A. Anal. Calcd for [Cu2(m-xpt)2(H2O)2](NO3)4.2H2O: C 42.76, H 3.59, N 22.66. Found: C 42.38, H 3.55, N 21.86. Cyclic voltammetry of complex 1 and complex 2 in DMF showed quasi-reversible waves at ca. 0.28 V vs. Ag/AgCl (−0.27 V for complex 1 and −0.28 V for complex 2 vs. Fc/Fc+). We investigated the reactivity of the Cu(I) dimers obtained by reducing complex 1 or 2. For chemical reduction of Cu(II) to Cu(I), we used sodium ascorbate to produce Cu(I) catalysts in situ for an azide-alkyne cyclization reaction. Treatment of complex 1 or 2 with sodium ascorbate in DMF under N2gave a yellow copper(I) complex. During the reduction of complex 1 or 2 to Cu(I), the Cu(II) d-d electronic absorption band disappeared, and an intense new band at 384 nm appeared ( Solutions of 3, generated by in situ reduction of complex 1 or 2 with sodium ascorbate in DMF, reacted with CO2(g) to produce the oxalate-bridged Cu(II) dimer [Cu2(m-xpt)2(μ-C2O4)](PF6)2, 4. The progress of the reaction was monitored by electronic absorption spectroscopy. We examined both the appearance of the Cu(II) d-d band in dimer 4 (λmax=751 nm, ε=98 M−1cm−1) and the disappearance of the Cu(I) metal-to-ligand charge transfer band at 384 nm, under CO2maintained at 1 atm. The reaction was nearly complete after 128 h. IR spectra of [Cu2(m-xpt)2(μ-C2O4)](PF6)2and [Cu2(m-xpt)2(μ-13C2O4)](PF6)2are shown in Intensity data were collected at low temperature on a Bruker Kappa Apex-II DUO CCD diffractometer fitted with an Oxford Cryostream chiller. MoKα (λ=0.71073 Å) radiation with a TRIUMPH curved graphite monochromator was used for 2, 4, and 4a. CuKα (λ=1.54184 Å) radiation from an I μS microfocus source with QUAZAR multilayer optics was used for 1 and 5. Data reduction included absorption corrections by a multiscan method, with SADABS, or TWINABS for 5. Structures were determined both by direct methods and by difference Fourier techniques, and were then refined by full-matrix least squares using SHELXL-97. All non-hydrogen atoms were refined anisotropically, except for the minor component in a disordered nitrate in 1 and 5, C atoms in 2, and the minor component of the disordered PF6− anion in 1 (vide infra). The crystal structure of dimer 4 contained dimeric cationic complexes with crystallographically imposed 2/m (C2h) symmetry. For 2, low data quality did not allow anisotropic refinement of the C atoms. Disordered solvents were removed using the SQUEEZE procedure for 2, resulting in non-integral solvent stoichiometry. Normal refinement procedures for 2 led to several unreasonable distances within the m-xpt ligands. Therefore, for the final refinement, the two sets of ligand atoms were restrained to yield similar bond distances and angles. For 4 and 4a, the PF6− anion was disordered in two orientations. A single solvent molecule (DMF for the crystals prepared by reaction of Cu(I) (3) with CO2; CH3CN for the crystals prepared by reaction of Cu(II) (1 or 2) with oxalate) was disordered across a mirror plane. The crystal of 5 was a nonmerohedral twin by 180° rotation about real axis [−1 0 1]. Refinement was versus HKLF5 data, and the twin components were present approximately 52/48%. This structure had a disorder involving a nitrate ion in two positions and a partially occupied water molecule associated with one of them. All H atoms were placed in idealized positions, except for water hydrogen atoms. Water H atoms were refined with restrained O—H distances, but the H atoms of the water molecule in the nitrate/water disorder of 5 could not be located. In one embodiment, a compound in accordance with the present invention is a binuclear compound having the structure [Cu2L2(μ-C2O4)p]Xnm+ wherein:
In another embodiment, a compound in accordance with the present invention is a binuclear compound having the structure [Cu2L2(μ-C2O4)p]Xnm+ wherein:
R1may be used to modify the properties of the internal cavity where CO2binds, or to control access to the cavity. R1may be, for example, H, C1-C4substituted or unsubstituted alkyl, —NH2, —OH, —CO2H;
The complete disclosures of all references cited in the specification are hereby incorporated by reference in their entirety, as is the complete disclosure of priority application Ser. No. 61/890,403. Also incorporated by reference is the complete disclosure of the following work by the present inventors, including its associated supplemental information: Pokharel, U. R., Fronczek, F. R. & Maverick, A. W. Cyclic pyridyltriazole-Cu(II) dimers as supramolecular hosts. Dalton Trans. 42, 14064-14067, doi:10.1039/C3dt52208c (2013). In the event of an otherwise irresolvable conflict, however, the disclosure of the present specification shall control. A composition and method are disclosed for capturing CO2. A pollutant/greenhouse gas can be converted into a valuable organic compound using a nontoxic reducing agent. CO2 may be economically captured from the atmosphere, from products of combustion, or from byproducts of various chemical processes. The method reduces CO2 in a three-step reaction cycle, in which a binuclear metal-organic compound converts CO2 to oxalate in a redox cycle under mild conditions. 1. A binuclear compound having the structure [Cu2L2(μ-C2O4)p]Xnm+ wherein:
p is 0 or 1; n is 0, 2, or 4; m is 0, 2, or 4; X is a monovalent anion; wherein if n is 2 or 4, the various X anions may be the same or different; and L is: wherein: R1is chosen from the group consisting of H, C1-C4substituted or unsubstituted alkyl, —NH2, —OH, and —CO2H; R2, R3, and R4are independently chosen from the group consisting of H, substituted or unsubstituted C1-C8alkyl, substituted or unsubstituted aryl, —OH, —OCnH2n−1, —O(CH2CH2O)kCH3, —O(CH2CH2O)kC2H5, N(CH3)3+, —SO3−, —NO2, and —CN; wherein k=1-8; R5, R6, R7, and R8are independently chosen from the group consisting of H, —OCH3, —N(CH3)2, halide, —NO2, —CN, substituted or unsubstituted C1-C8alkyl, substituted or unsubstituted aryl, —OH, —OCjH2j−1, —O(CH2CH2O)jCH3, —O(CH2CH2O)jC2H5, —N(CH3)3+, —SO3−, —NO2, and —CN (j=1-8). 2. The compound of 3. The compound of 4. The compound of 5. A method for reducing carbon dioxide to oxalate; said method comprising the sequential steps of:
(a) reacting carbon dioxide with a reduced form of the compound of (b) reacting the oxalate complex from step (a) with a mineral acid to liberate oxalate from the oxidized form of the compound of (c) reducing the oxidized form of the compound of (d) repeating each of steps (a) through (c) a plurality of times. 6. A method for reducing carbon dioxide to oxalate; said method comprising the sequential steps of:
(a) reacting carbon dioxide with a reduced form of the compound of (b) reacting the oxalate complex from step (a) with a mineral acid to liberate oxalate from the oxidized form of the compound of (c) reducing the oxidized form of the compound of (d) repeating each of steps (a) through (c) a plurality of times. 7. A method for reducing carbon dioxide to oxalate; said method comprising the sequential steps of:
(a) reacting carbon dioxide with a reduced form of the compound of (b) reacting the oxalate complex from step (a) with a mineral acid to liberate oxalate from the oxidized form of the compound of (c) reducing the oxidized form of the compound of (d) repeating each of steps (a) through (c) a plurality of times. 8. A method for reducing carbon dioxide to oxalate; said method comprising the sequential steps of:
(a) reacting carbon dioxide with a reduced form of the compound of (b) reacting the oxalate complex from step (a) with a mineral acid to liberate oxalate from the oxidized form of the compound of (c) reducing the oxidized form of the compound of (d) repeating each of steps (a) through (c) a plurality of times.TECHNICAL FIELD
BACKGROUND ART
DISCLOSURE OF THE INVENTION
BRIEF DESCRIPTION OF THE DRAWINGS
METHODS
EXAMPLE 1
Equipment and Materials
EXAMPLE 2
[Cu2(m-xpt)2(NO3)2](PF6)2, 1
EXAMPLE 3
[Cu2(m-xpt)2Cl2](PF6)2, 2
EXAMPLE 4
[Cu2(m-xpt)2](PF6)2, 3
EXAMPLE 5
[Cu2(m-xpt)2(μ-C2O4)](PF6)2, 4
EXAMPLE 6
[Cu2(m-xpt)2(μ-13C2O4)](PF6)2
EXAMPLE 7
[Cu2(m-xpt)2(μ-C2O4)](PF6)2, 4a
EXAMPLE 8
[Cu2(m-xpt)2(H2O)2](NO3)4, 5
Results
EXAMPLE 9
Cyclic Voltammetry
EXAMPLE 10
IR Spectra
EXAMPLE 11
X-ray Data Collection and Structure Determination
EXAMPLE 12
Crystal Data and Structure Refinement Parameters
Crystal Data and Structure Refinement Parameters for 1 and 2. Compound 1 2 deposition no. CCDC 1000457 CCDC 1000458 formula [Cu2(m-xpt)2(NO3)2](PF6)2· [Cu2(m-xpt)2Cl2](PF6)2· 3.5CH3CN 4.44C3H7NO M 1329.93 1601.17 crystal system Monoclinic Orthorhombic space group P21/c Pna21 a/Å 11.4090(16) 13.810(2) b/Å 12.8455(18) 19.380(3) c/Å 23.411(3) 24.711(3) β/deg 108.575(5) 90 V/Å3 3252.3(8) 6613.6(16) Z 2 4 T/K 100.0(5) 90.0(5) Dcalc/g cm−3 1.358 1.608 crystal 0.35 × 0.17 × 0.10 0.04 × 0.14 × 0.31 dimensions/mm Radiation CuKα MoKα θ limits/deg 3.98-59.20 1.65-25.71 reflns, measd/ 22792/4639/3982 52940/12074/7203 unique/obsd F(000) 1340 3278 μ/mm−1 2.066 0.871 Rint 0.0468 0.0525 R[I>2σ(I)] 0.1013 0.0830 RW(all data) 0.3540 0.2384 GOF 1.624 1.053 Crystal Data and Structure Refinement Parameters for 4, 4a, and 5. Compound 4 4a 5 deposition no. CCDC 984468 CCDC 984469 CCDC 984470 formula [C46H36Cu2N16O4](PF6)2· [C46H36Cu2N16O4](PF6)2· [C44H40Cu2N16O2](NO3)4· 2C3H7NO 4CH3CN 2C3H7NO · 3.34H2O M 1440.12 1458.14 1406.50 crystal Orthorhombic Orthorhombic Monoclinic system space group Cmca Cmca P21/n a/Å 24.1471(17) 24.461(7) 11.4361(13) b/Å 11.7107(7) 11.959(3) 22.691(3) c/Å 20.5949(12) 20.008(5) 11.9848(13) β/deg 90 90 106.847(5) V/Å3 5823.8(6) 5853(3) 2976.6(6) Z 4 4 2 T/K 100.0(5) 100.0(5) 100.0(5) Dcalc/g cm−3 1.642 1.655 1.569 Crystal 0.45 × 0.17 × 0.04 0.30 × 0.22 × 0.13 0.12 × 0.19 × 0.21 dimensions mm radiation MoKα MoKα CuKα θ limits/deg 1.69-30.05 2.04-30.23 4.32-69.67 reflns, measd/ 44218/4357/3592 31270/4418/3607 52513/14803/12358 unique/obsd F(000) 2928 2960 1455 μ/mm−1 0.891 0.886 1.691 Rint 0.0310 0.0447 0.0741 R[I>2σ(I)] 0.0527 0.0346 0.0637 RW(all data) 0.1531 0.0900 0.1928 GOF 1.033 1.031 1.024
R2, R3, and R4, particularly R3, may be used to enhance the solubility of the complexes in certain solvents. For example, R3═OCH2CH2CH2CH3is expected to increase solubility in nonpolar solvents such as toluene. R2, R3, and R4may, for example, be independently chosen from H, substituted or unsubstituted C1-C8alkyl, substituted or unsubstituted aryl, —OH, —OCkH2k−1, —O(CH2CH2O)kCH3, —O(CH2CH2O)kC2H5
—N(CH3)3+, —SO3−, —NO2, —CN (k=1-8);
R5, R6, R7, and R8may be used to modify solubility, or to modify the electron-donating ability of the ligand as it coordinates to the Cu ions, or both. Modifying the electron-donating ability may be useful in altering the reducing strength of the Cu(I) complexes, which will in turn affect the rate of reaction of Cu(I) with CO2. R5, R6, R7, and R8may, for example, be independently chosen from: H;
—OCH3, —N(CH3)2(electron-releasing);
halide, —NO2, —CN (electron-withdrawing); or
substituted or unsubstituted C1-C8alkyl, substituted or unsubstituted aryl, —OH, —OCjH2j−1, —O(CH2CH2)jCH3, —O(CH2CH2O)jC2H5, —N(CH3)3+, —SO3−, (j=1-8).