METHOD
The present invention relates to a method for producing leafy biomass in culture. The listing or discussion of an apparently prior-published document in this specification should not necessarily be taken as an acknowledgment that the document is part of the state of the art or is common general knowledge. The production of biomass in culture is useful for the production of genetically engineered polypeptides, for the production of endogenous plant products, including medicinal products, polysaccharides, lignins and lipids, for the production of novel simple and complex chemicals not naturally found in plants through metabolite engineering, including new forms of polysaccharides, lignins, sugars, aromatic and aliphatic compounds, and for capturing carbon dioxide. The biomass may also be used for fuel in some circumstances. WO 00/57690 relates to the micropropagation and production of phytopharmaceutical plants from differentiated plant pieces. In particular WO 00/57690 relates to the stimulation of small pieces of differentiated cells taken from an adult plant to produce new plantlets which can be grown to fully-formed phytopharmaceutical-producing plants capable of growth normal plant growth in typical plant growth media (e.g. soil, compost). WO 01/94602 relates to a method for regenerating plants and uses thereof to multiply and/or transform plants using solid growth media. The plants resulting from the methods described in WO 01/94602 are viable plants that may grow under normal growth in typical plant growth media (e.g. soil and compost). WO 2008/028115 relates to high-throughput methods for producing large numbers of transgenic corn plants in a short space of time by the use of a single container system for transgenesis, and growth into a viable plant. The corn plants produced are viable plants with root, stem and leaf structures and that are capable of normal plant growth in typical plant growth media (e.g. soil, compost). The use of temporary liquid immersion culture systems (e.g. temporary immersion bioreactors or TIBs) is known, for example from Etienne & Berthouly (2002) One of the main reasons why researchers have chosen plants for expressing biopharmaceuticals and other high-value proteins is the formidable scale-up possibility and very low maintenance costs that are associated with plant growth. However, the use of transgenic plants has its drawbacks, with public concern about the transfer of transgenes to surrounding non-transgenic crops and the possibility of food chain contamination (Fox, 2003). For a long time, it was assumed that the plastid genome in most species was absent in pollen and was inherited maternally (Hagemann, 2004; Zhang et al, 2003; Scott and Wilkinson, 1999). Consequently, insertion of genes into the chloroplast genome, or plastome, to generate transplastomic plants, was considered to provide an intrinsic natural barrier to the pollen-mediated flow of transgenes. However, several recent publications have shown that the leak in chloroplast DNA containment is more frequent and widespread than originally thought. For example, transfer of chloroplast DNA to the pollen was estimated to reach 0.03% in Another concern is the possibility of chloroplast DNA being transferred to the nuclear genome over time (Sheppard et al, 2008), from where it could be passed on to a nearby non-transgenic species, in the same way as for a classic nuclear transformant. A frequency of one chloroplast DNA transfer to the nuclear DNA in every 16,000 pollen grains was detected in tobacco (Huang et al, 2003). Taking into account the fact that between 5,000 to 16,000 tobacco plants can be grown per acre, depending on the tobacco species, the risk of chloroplast DNA transfer to the nucleus is not negligible. Concerns have also been raised that antibiotic-resistance cassettes, such as the aadA gene, which is used to select for chloroplast transformants, could be transferred to soil bacteria (Monier et al, 2007) and bacteria found in the gut of feeding insects (Brinkmann and Tebbe, 2007). To circumvent any environmental issues that could result from planting transplastomic seeds in the field, one solution is to produce recombinant proteins in plant cell suspension cultures grown under contained conditions. Indeed, plant cell suspensions have been modified to express a large number of heterologous proteins (reviewed in Hellwig at al, 2004). Plant cell suspensions display several advantages over whole plants for the production of recombinant proteins, such as a shorter period of time before harvest, fully controlled growth and independence from weather conditions or diseases. Current good manufacturing practices, cGMP, based on bacterial production systems, can also be applied easily, leading to a quicker regulatory approval by the Federal Drug Administration (FDA) or by the European Agency for the Evaluation of Medicinal Products (EMEA) (reviewed in Ma et al, 2003; Fischer et al, 2004; Twyman et al, 2003). Like bacteria, plant cell suspension cultures are inexpensive to grow and maintain. They are also intrinsically safe, because they neither harbour human pathogens nor produce endotoxins. Plant cell suspensions can be maintained in simple, synthetic media, but can synthesize complex multimeric proteins just like animal cells. In contrast to field-grown plants, the performance of cultured plant cells is independent of the climate, soil quality, season and day length. There is no risk of contamination with mycotoxins, herbicides or pesticides (Doran, 2000) and there are fewer by-products (e.g. fibres, oils, waxes, phenolic compounds). Perhaps the most important advantage of plant cell suspension cultures over whole plants is the much simpler procedures for product isolation and purification (Fischer et al, 1999). However, the main disadvantages of plant cell suspension cultures are the slow growth and the usually low yields of recombinant protein produced by nuclear transformation (Hellwig et al, 2004). Another weakness is the fact that the productivity of plant cell cultures can vary considerably, with recombinant protein levels usually ranging from 0.0064% to 4% of total soluble protein (TSP), although in exceptional cases up to 20% of TSP can be achieved (Huang et al, 2001). In general, chloroplast transformation better yields of recombinant protein than classic nuclear transformation. For example, the B-subunit of In the work described in the Examples, the expression levels of a plastid-encoded recombinant protein have been investigated, in this case a variant of the Green Fluorescent Protein GFP+ (Scholz et al, 2000) in leaf tissue, callus and cell suspensions grown under various conditions. The results indicate that expression in cell suspension cultures is a feasible route for high-level and contained expression of a foreign protein in the chloroplast although levels of expression are much less than that in plant leaves. There is also described the development a new expression system, based on temporary immersion bioreactors, which is able to produce extremely high-levels of recombinant protein starting from cell suspension cultures, and able to produce of high levels of leafy biomass from undifferentiated plant cells. A first aspect of the invention provides a method for producing leafy biomass from undifferentiated plant cells, the method comprising providing undifferentiated plant cells, contacting them with an agent that promotes differentiation of the cells into leafy tissue and growing the cells in a temporary liquid immersion culture system. By “undifferentiated plant cells” we include the meaning that the cells show substantially no signs of being differentiated into any particular plant tissue such as shoot or leaf, and that they will remain in that state for at least one month under conditions where no agent which induces differentiation of undifferentiated cells is present, in particular there should be no agent that induces differentiation of undifferentiated cells into shoots. The undifferentiated cells may be transgenic or non-transgenic. Typically, the undifferentiated cells can be derived from a permanent callus or callus material. A permanent callus is a cell culture of undifferentiated plant cells. Such permanent callus cells remain in an undifferentiated form for at least one month. Undifferentiated cells can also be derived in-vitro from differentiated plant material, such as leaves, stems, flowers, seeds or roots, which are cut and placed in contact with certain plant hormones, such as Auxins. When this plant material has been in contact with the hormones, calli will form in some areas of the plant material. The calli that are induced by hormones on differentiated plant material are not considered to be a permanent callus. The step of providing the undifferentiated cells where the plant is not a transgenic plant comprises:
The cut plant material may be all or part of a root, leaf, stem, flower or seed. The step of providing the undifferentiated cells where the plant is a transplastomic plant comprises:
The callus of the current invention work is a permanent callus, having been cultivated and maintained for at least one month as undifferentiated cells Preferably, the only cells that are present when contacting with the agent are undifferentiated cells. Typically, at least 90%, or 95%, or 99%, or 99.9% or 99.99% of the cells present when contacting with the agent are undifferentiated cells. Preferably, substantially all leafy and leaf like biomass material is produced upon differentiation of the undifferentiated cells following contact with the agent. Typically, the plant material produced upon treatment of the undifferentiated cells with the agent should be at least 50% leafy biomass, preferably 70%, and more preferably greater than 85%. By “leafy” and “leaf like” biomass” we include the meaning that the plant material is in the form of leaf or “leaf like” tissue. These leafy tissues are distinguished from other plant tissue by the shape of the tissue pieces, the number of chloroplasts and the significant photosynthetic activity. For example, for any given plant, leaf material has a higher number of chloroplasts and developing chloroplasts, as counted by confocal microscopy analysis of the plant tissue, and these chloroplasts have higher photosynthetic activity (determination of Fv/Fm with fluorometer) and higher chlorophyll content (by analysis of extracted pigments by absorption spectrophotometry) than chloroplasts in non-leaf material, as detected by the absorption of carbon dioxide by the plant tissue. Such methods of determination are well known to the skilled person as for example as described in (Baker (2008) The temporary liquid immersion culture system may be any such system as are known in the art (for example see Etienne & Berthouly (2002) The plant cells may be cells from a monocotyledon or a dicotyledon. Suitable dicotyledon plants include any of a tobacco, potato, tomato, bean, soybean, carrot, cassava, or Suitable monocotyledon plants include any of corn, rye, oat, millet, sugar cane, sorghum, maize, wheat or rice. In a preferred embodiment, the plant cells are from a medicinal plant in which the main medicinal product is produced in the leaves. It will be appreciated that the method represents an advantageous approach to obtaining such medicinal products by extracting them from the leafy biomass. Suitable medicinal plants include any of The medicinal compounds produced by the leafy biomass may be incorporated into pharmaceutical compositions by combination with pharmaceutically acceptable excipients, diluents or carriers. In a further preferred embodiment, the plant may be an energy crop. By energy plants, we mean plant species used in the production of biofuels including ethanol or biodiesel. The current invention allows for a continuous production of biomass that can be employed for a continuous production of biofuel, independent from the season and plant species. The biomass generated can endogenously contain relatively elevated levels of polysaccharides, for use in fermentation based ethanol production processes, or relatively high levels of one or more lipids that can be further processed for the production of biodiesel. These elevated levels of advantageous compounds can also be generated in the biomass by genetic engineering. Suitably, the plant is any of The agent that promotes differentiation of the cells into leafy tissue is typically a plant hormone (phytohormone or plant growth substance), and preferably a cytokinin. Cytokinins are a group of chemicals that primarily influence cell division and shoot formation but also have roles in delaying cell senescence, are responsible for mediating auxin transport throughout the plant, and affect internodal length and leaf growth. Auxins are compounds that positively influence cell enlargement, bud formation and root initiation. They also promote the production of other hormones and in conjunction with cytokinins, they control the growth of stems, roots, fruits and convert stems into flowers. The cytokinin may be any natural or artificial cytokinin belonging to the adenine-type or the phenylurea-type. Preferably, the cytokinin is any of adenine, kinetin, zeatin, 6-benzylaminopurine, diphenylurea, thidiazuron (TDZ) and their respective derivatives which have cytokinin activity The agents may promote, induce, and provoke differentiation such that shoots grow rapidly, preferably in an exponential manner, from any single undifferentiated plant cells derived from callus/cell suspension of the invention. Such shoots develop into leafy or leaf like biomass. Preferably the agent that promotes differentiation of the cells into leafy tissue is thidiazuron (TDZ). Conveniently, the agent may be used in combination with another plant hormone, such as an auxin, such as the naturally occurring auxins, 4-chloro-indoleacetic acid, phenylacetic acid (PAA), indole-3-butyric acid and indole-3-acetic acid; or the synthetic auxin analogues 1-naphthaleneacetic acid (NAA), 2,4-dichlorophenoxyacetic acid. Typically, the agent is added in the culture medium at a concentration of from 0.01 to 100 μM. Preferably the concentration is between 0.1 and 10 μM. The agent may be added at the start of or during the temporary liquid immersion culture step. Any suitable immersion regime may be selected, for example to optimise the production of leafy biomass or to optimise the concentration of a particular product in the leafy biomass, such as a polypeptide or medicinal product of interest. Typically, the immersion time varies from 1 to 30 minutes every 2 to 24 hours of culture. Preferably, the immersion time is between 1 and 10 minutes every 2 to 6 hours. The skilled person will readily be able to select the most appropriate immersion culture parameters such as time, temperature and growth media based on the plant species and origin in order to generate a specific biomass for a specific purpose in the most effective manner i.e. at the most appropriate speed, quantity and quality. The volume of liquid in the temporary liquid immersion culture may be any convenient volume but typically is from 1 to 10,000 litres. Alternatively, the volume may be between 1 and 5,000 litres, 1 and 1,000 litres, or 1 and 500 litres. The vessel containing the temporary liquid immersion culture system may be any convenient size, and typically is from 1 to 10,000 litres. Alternatively, the volume may be between 1 and 5,000 litres, 1 and 1,000 litres, or 1 and 500 litres. In one embodiment of the invention, the plant cells are not genetically engineered. As is well know, plants produce endogenously many important products in their leaves such as medicinal products as described above, as well as oils, pigments, antioxidants, simple and complex biochemicals such as sugars (carbohydrates), lipids, amino acids, volatile aromatic compounds, and flavours/flavour precursors. The plant material of interest may also be capable of concentrating, capturing, or degrading, toxic pollutants in a sample, such as in a feed water source (plant based in-vitro decontamination/purification). The plant material may also be used to transform one compound contained in the temporary reaction solution into one or more other compounds. In another embodiment of the invention, the plant cells are genetically engineered, for example to express a polypeptide. The polypeptide may be any polypeptide of interest, but preferably is any one of a therapeutic polypeptide, an enzyme, a growth factor, an immunoglobulin, a hormone, a structural protein, a protein involved in stress responses of a plant, a biopharmaceutical, a peptide, or a vaccine antigen. When the polypeptide is an enzyme it may be used to alter the metabolism of the leafy material, thereby allowing the generation of novel polymers and metabolites. One or more polypeptides may also be expressed inside the leafy material to amplify the ability of the leafy tissue to purify or degrade pollutants found in a sample, such as a water source. The genetically engineered plant cell (recombinant or transgenic plant cell) may be (i) a nuclear transformed plant cell in which the exogenous nucleic acid (transgene) resides in the nucleus; (ii) a transplastomic plant cell in which the exogenous nucleic acid (transgene) resides in a plastid, such as a chloroplast; or (iil) a plant cell that is both nuclear transformed and transplastomic. Methods of making nuclear transformed plants and transplastomic plants are well known in the art. For example, nucleic acid molecules may be introduced into plant cells using particle bombardment, micro-injection, PEG-electroporation, It is preferred if the plant is a transplastomic plant. Plants may be transformed in a number of art-recognised ways. Those skilled in the art will appreciate that the choice of method might depend on the type of plant targeted for transformation. Examples of suitable methods of transforming plant cells include microinjection (Crossway et al., Successfully transformed cells, i.e. cells that contain a DNA construct of the present invention, can be identified by well known techniques. For example, one selection technique involves incorporating into the expression vector a DNA sequence (marker) that codes for a selectable trait in the transformed cell. These markers include dihydrofolate reductase, G418 or neomycin resistance for eukaryotic cell culture, and tetracyclin, kanamycin or ampicillin resistance genes for culturing in The marker gene can be use to identify transformants but it is desirable to determine which of the cells contain recombinant DNA molecules and which contain self-ligated vector molecules. This can be achieved by using a cloning vector where insertion of a DNA fragment destroys the integrity of one of the genes present on the molecule. Recombinants can therefore be identified because of loss of function of that gene. Another method of identifying successfully transformed cells involves growing the cells resulting form the introduction of an expression construct of the present invention to produce the polypeptide of the invention. Cells can be harvested and lysed and their DNA content examined for the presence of the DNA using a method such as that described by Southern (1975) In addition to directly assaying for the presence of recombinant DNA, successful transformation can be confirmed by well known immunological methods when the recombinant DNA is capable of directing the expression of the protein. For example, cells successfully transformed with an expression vector produce proteins displaying appropriate antigenicity. Samples of cells suspected of being transformed are harvested and assayed for the protein using suitable antibodies. Those skilled in the art will appreciate that stable and unstable (transient) transformants may be produced by plant transformation techniques. Transient transformants only transiently express the product comprising the compound of the invention encoded by the DNA construct. Transient expression systems can be useful for molecular genetic studies as well as for some specific commercial applications wherein the transformed cells that are responsible for the production of a valuable protein are harvested shortly after the transformation. Stable transformants may be produced when the heterologous DNA sequence integrates into the genome of the host. With regard to plants the heterologous DNA may be inserted into one of the chromosomes or into the organelle genomes (mitochondrion, chloroplast). Those skilled in the art will appreciate that Examples of vectors include cloning vectors, expression vectors and shuttle vectors. Cloning vectors include agents that are used to carry the fragment of DNA into a recipient for the purposes of producing more of a DNA sequence. Expression vectors include agents that carry the DNA sequence into a host and directs therein the synthesis of a specific product, such as a protein or antisense transcript. An expression vector may be produced by insertion of the coding DNA sequence into an expression cassette containing an insertion site in the vector. Shuttle vectors include a genetic element that is constructed to have origins of replication for two hosts so that it can be used to carry a foreign sequence to more than one host. For example, the shuttle vector may have origins of replication for Generally, the DNA is inserted into a vector in proper orientation and correct reading frame for expression. If necessary, the DNA may be linked to the appropriate trancriptional and translational regulatory control nucleotide sequences recognised by the desired host, although such controls are generally available in the vector. Regulatory elements may be derived from a plant or from an alternative source, including plant viruses or the Ti/Ri plasmid of The DNA insert may be operatively linked to an appropriate promoter, for example a plant viral promoter or a plant promoter. Preferable promoters include constitutive, inducible, temporally regulated, developmentally regulated, cell-preferred and/or cell-specific promoters, tissue-preferred and/or tissue-specific promoters, and chemically regulated promoters. The promoter may also be a synthetic or artificial promoter constructed from artificial combinations of transcription factor binding sites. Constitutive promoters include the CaMV 35S and 19S promoters (Fraley et al., U.S. Pat. No. 5,352,605). The promoter expression cassettes described by McElroy et al., Yet another preferred constitutive promoter is derived from ubiquitin, which is another gene product known to accumulate in many cell types. The ubiquitin promoter has been cloned from several species for use in transgenic plants (e.g. Binet et al., Inducible promoters include promoters which are responsive to abiotic and biotic environmental stimuli. Abiotic environmental stimuli include light, temperature and water availability. Biotic environmental stimuli include pathogens, (including viral induced, bacterial induced, fungal induced, insect induced, and nematode induced promoters), interactions with symbionts and herbivores. Promoters may also be responsive to movement, touch, tissue damage and phytohormones (including abscissic acid, cytokinins, auxins, giberellins, ethylene, brassinosteroids and peptides such as systemin and nodulation factors). Temporally regulated promoters include circadian regulated promoters as well as those which respond to non-circadian time-keeping mechanisms. Developmentally regulated promoters include tissue specific and cell type specific promoters for organs and other structures, including leaves, stems, roots, flowers, seeds, embryos, pollen and ovules. Tissue-specific or tissue-preferential promoters useful for the expression of the coding sequence in plants, particularly maize and sugar beet, are those which direct expression in root, pith, leaf or pollen. Examples are the TUB1 promoter from Particularly preferred is the 16S rRNA, psbA and rbcL promoter. In addition to promoters, a variety of transcriptional terminators may be incorporated into the DNA constructs of the present invention. Transcriptional terminators are responsible for the termination of transcription beyond the transgene and its correct polyadenylation. The transcriptional terminator may be derived from the same gene as the promoter or may be derived from a different gene. In a preferred embodiment, the coding sequence is operably linked to its naturally occurring polyadenylation signal sequence. Appropriate transcriptional terminators and those which are known to function in plants include the CaMV 35S terminator, the tml terminator, the pea rbcS E9 terminator and others known in the art. Convenient termination regions are also available from the Ti-plasmid of In addition to the above, the DNA construct of the present invention may comprise any other sequence that can modulate expression levels. Numerous sequences have been found to enhance gene expression from within the transcriptional unit and these sequences can be used in conjunction with a coding sequence to increase expression in transgenic plants. Various intron sequences have been shown to enhance expression, particularly in monocotyledonous cells. For example, the introns of the maize Adh1 gene have been found to significantly enhance the expression of the wild-type gene under its cognate promoter when introduced into maize cells (Callis at al, The constructs can also include a regulator such as a chloroplast localisation signal, chloroplast specific promoters, chloroplast specifc sequence homologues to drive homologous recombination, nuclear localization signals (Lassner et al., Plant transformation vectors commonly used are The method of the first aspect of the invention may be used to capture carbon dioxide. Air can be used for this, although it is preferred if the air is enriched with carbon dioxide, for example it may contain up to 10% carbon dioxide. In addition, to allowing for more efficient carbon dioxide capture, it will allow for further production of biomass by virtue of additional carbon being made available to the plant cells. Carbon dioxide capture can be achieved by providing air containing carbon dioxide to the temporary immersion bioreactor. The source of carbon dioxide may be from any source including atmospheric carbon dioxide, a carbon dioxide canister, the exhaust gas of a power plant or the exhaust gas of a combustion and/or a fermentation chamber. The carbon dioxide concentration may advantageously be controlled in order to regulate the pH of growth medium and the leafy biomass growth in the temporary immersion bioreactor. Biofuels may be produced by a method having the steps of: growing the leafy biomass in the temporary immersion bioreactors described above, for example the bioreactor being one or more closed temporary immersion bioreactors; harvesting the leafy biomass in a continuous, semi-continuous or batch mode process; and converting lipids or carbohydrates from the leafy biomass into a biofuel. The lipids or carbohydrates may be extracted from the leafy biomass either before or as part of the process of conversion into biofuel. The lipids or carbohydrates may alternatively be secreted into the culture medium by the leafy biomass and harvested from the culture medium for conversion to a biofuel. In order to improve the production of biofuel by the leafy biomass, the biomass may be subjected to an environmental stress, or a combination of several stresses, to increase lipid and or carbohydrate production. The leafy biomass may also be genetically engineered in order to improve the production and accessiblity (e.g. by promoting secretion into the culture medium) of the lipid or carbohydrate that will be converted to biofuel. Biodiesel may be produced from oils/lipids by the process of transesterification and is a liquid similar in composition to fossil/mineral diesel. Its chemical name is fatty acid methyl (or ethyl) ester (FAME). Oils are mixed with sodium hydroxide and methanol (or ethanol) and the chemical reaction produces biodiesel (FAME) and glycerol. Bioalcohol compounds are biologically produced alcohols, most commonly ethanol (bioethanol), and less commonly propanol and butanol, and are produced by the action of microorganisms and enzymes through the fermentation of sugars, starches, or cellulose. A second aspect of the invention provides a method of producing a polypeptide in plant cells in vitro comprising:
In other words, the cells are propagated by a method comprising providing undifferentiated plant cells, contacting them with an agent that promotes differentiation of the cells into leafy tissue and growing the cells in a temporary liquid immersion culture system. Transgenic nucleic acid molecules can be introduced into chloroplasts using methods described above and in the Examples. By homoplastomy we mean the situation where most or all of the multiple copies of the chloroplast DNA in each chloroplast of a plant cell are transformed. Homoplastomy is achieved by subculturing the transplastomic material several times, on media containing a selective agent. The selective agent is associated with a selectable marker used in the transformation construct, and can be any appropriate selectable marker, for example a resistance gene for antibiotics, such as spectinomycin or kanamycin. Achievement of homoplastomy is standardly verified using Southern blotting. The step of providing the undifferentiated cells where the plant is not a transgenic plant comprises:
The cut plant material may be all or part of a root, leaf, stem, flower or seed. The step of providing the undifferentiated cells where the plant is a transplastomic plant comprises:
The transgenic construct should contain at two least nucleic acid sequences similar (e.g. above 85% identity) to the targeted chloroplast DNA so as to achieve homologous recombination (the so-called right and left borders); a selectable marker gene and an encoded peptide or polypeptide sequence; Regarding the use of transplastomic undifferentiated cells, homoplastomy is achieved using antibiotic selection, for example selection with, streptomycin spectinomycin or kanamycin. Callus homoplastomy can be achieved by various methods well known to the skilled person including, but not limited to:
Preferably, the nucleic acid molecule comprises a selectable marker gene. Typically, the selectable marker gene is an antibiotic resistance gene such as aadA, nptII, AphVI. Typically, the nucleic acid molecule is inserted into a vector or a PCR fragment. Typically, the vector is a plasmid, and typically it can be propagated in It is preferred if the expression of the polypeptide is driven by a strong chloroplast specific promoter. Suitable promoters include a 16S rRNA promoter, a psbA promoter and a rbcL promter. Methods of plant cell and chloroplast transformation are well know to the skilled person and include transgenic methods as discussed above and as described in Sambrook and Russell (2001), Molecular Cloning, A laboratory manual; Grierson and Covey (1988) Plant molecular biology and Watson et al. (1997) Recombinant DNA. The amount of light available and/or the amount of sucrose available in the growth medium may influence the production of the polypeptide. The growth media and conditions including the gas mixture (e.g. carbon dioxide concentration) can be readily optimised by a skilled person for the production of each specific polypeptide based on the plant material being used and the biomass required to be produced. The method of the second aspect of the invention preferably includes the further step of obtaining the polypeptide from the leafy biomass. The polypeptide so-obtained is also included within the invention. Conveniently, the polypeptide is obtained by crushing the leafy tissue to produce a tissue extract and isolating the polypeptide from the tissue extract. Conveniently, the polypeptide is purified from the tissue extract using at least one of filtration, HPLC, ion exchange resin extraction, hydrophobic interaction resin extraction, affinity chromatography or oil-water phase separation. The polypeptide may comprise a tag for use in purifying the polypeptide. The tag may be a cleavable or non-cleavable tag, such as any one of a GST, biotin, 6His, Strep, HA or myc tag. The invention also includes leafy biomass obtained by method of the first aspect of the invention. The polypeptide obtained from the method may be any one of a therapeutic polypeptide, an enzyme, a growth factor, an immunoglobulin, a hormone, a structural protein, a protein involved in stress responses of a plant, a biopharmaceutical or a vaccine antigen A third aspect of the invention provides a method for obtaining a component present in leafy biomass, the method comprising producing leafy biomass according to the first aspect of the invention and obtaining the component from the leafy biomass. Typically, the component is obtained in a substantially pure form, and so the method may comprise the further step of purifying the component. The substantially pure form typically contains >90%, or >95% or >99% of the component. The component may be obtained by its secretion from the leafy biomass or by extraction from the leafy biomass, for example by crushing the leafy biomass to release the component. The component obtained may be a medicinal product, a recombinantly expressed polypeptide, a carbohydrate, a lipid, an oil, a volatile aromatic compound, an anti-oxidants, a pigment, a flavour or flavour precursor; and the component may be either endogenous or exogenous. The invention further provides for the processing of the component obtained into a further product, for example a biofuel, food stuff or medicinal product. The invention also includes a system for producing a polypeptide in plant cells in vitro comprising: an agent which promotes differentiation of undifferentiated cells into leafy tissue; and a
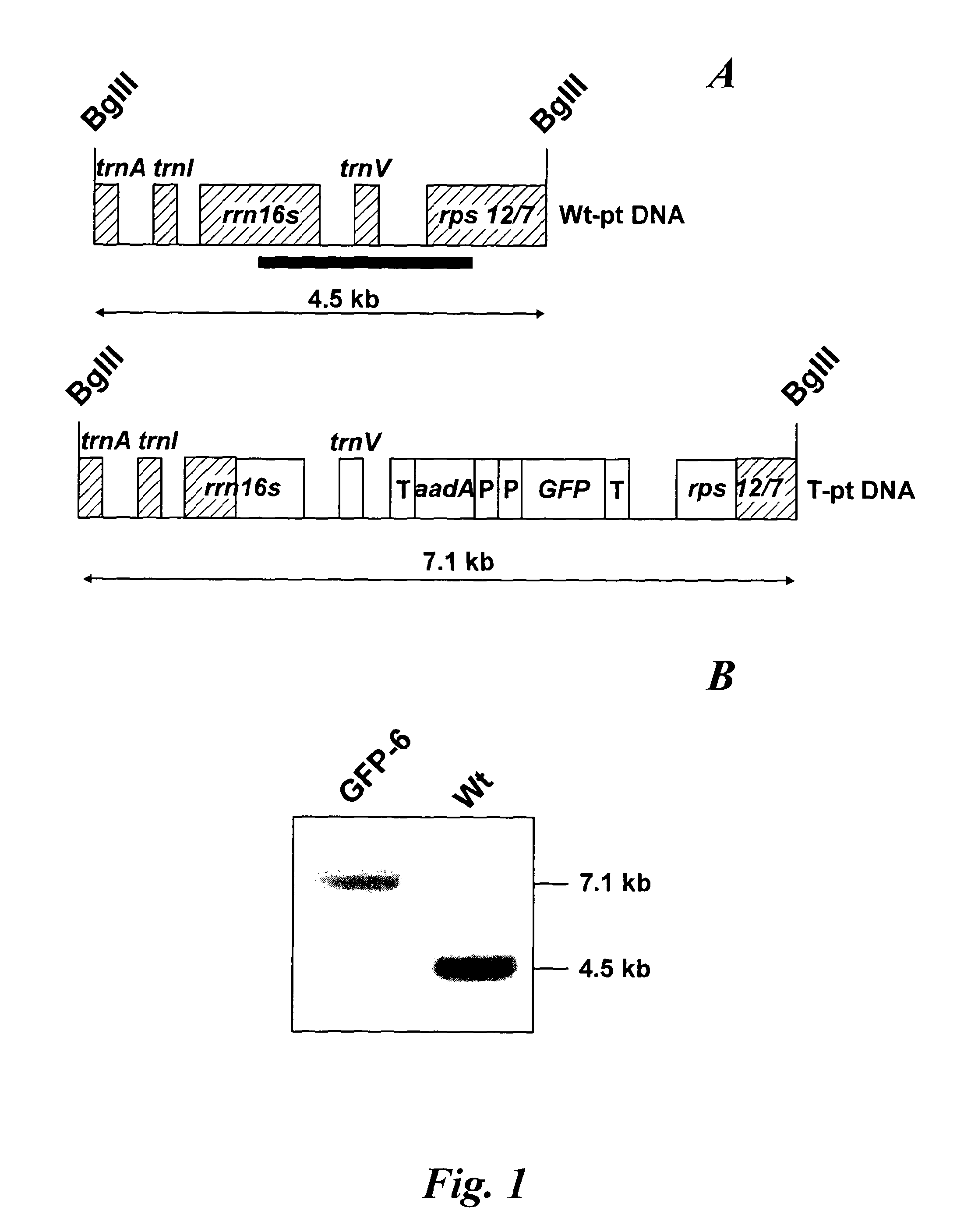
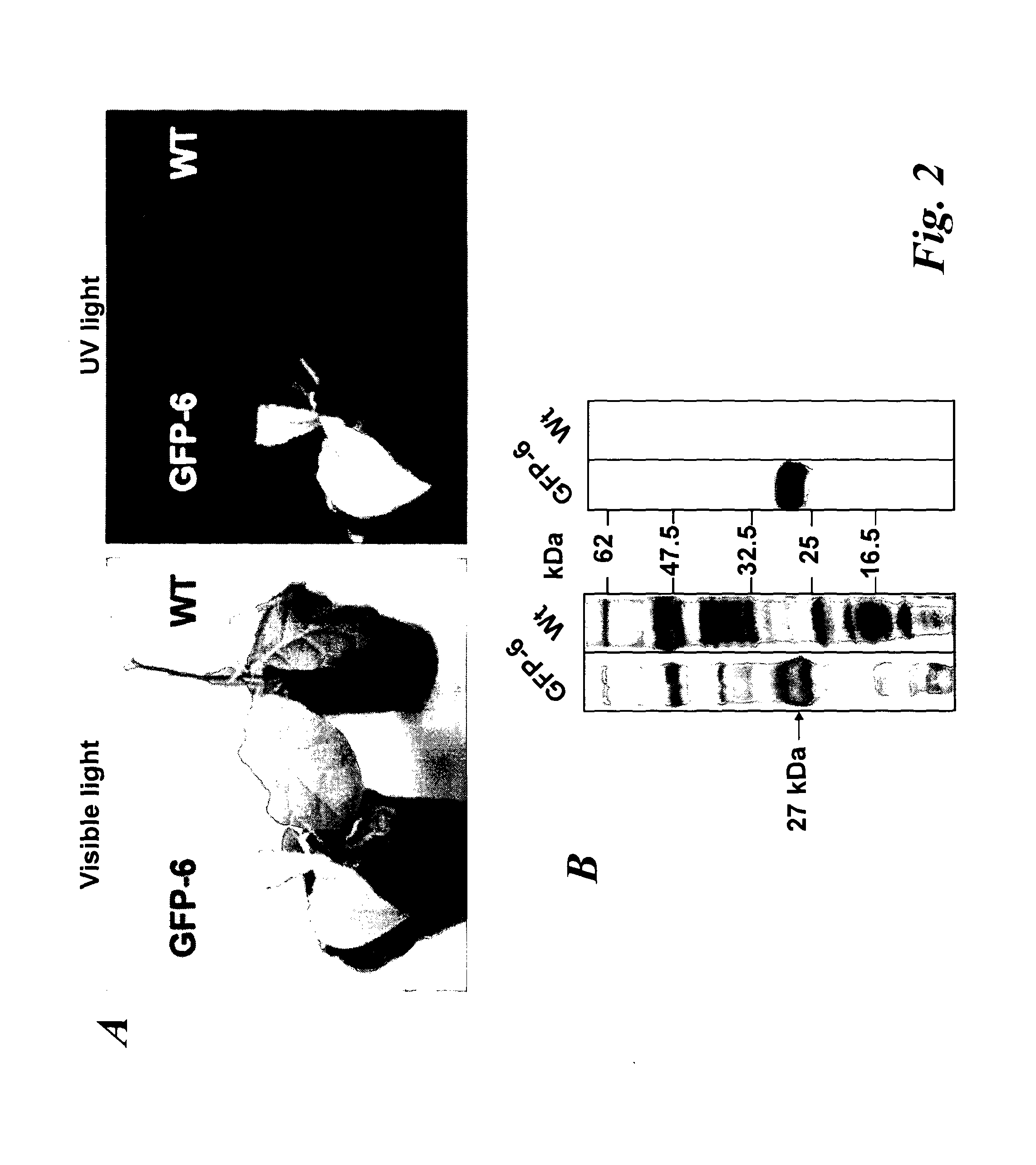
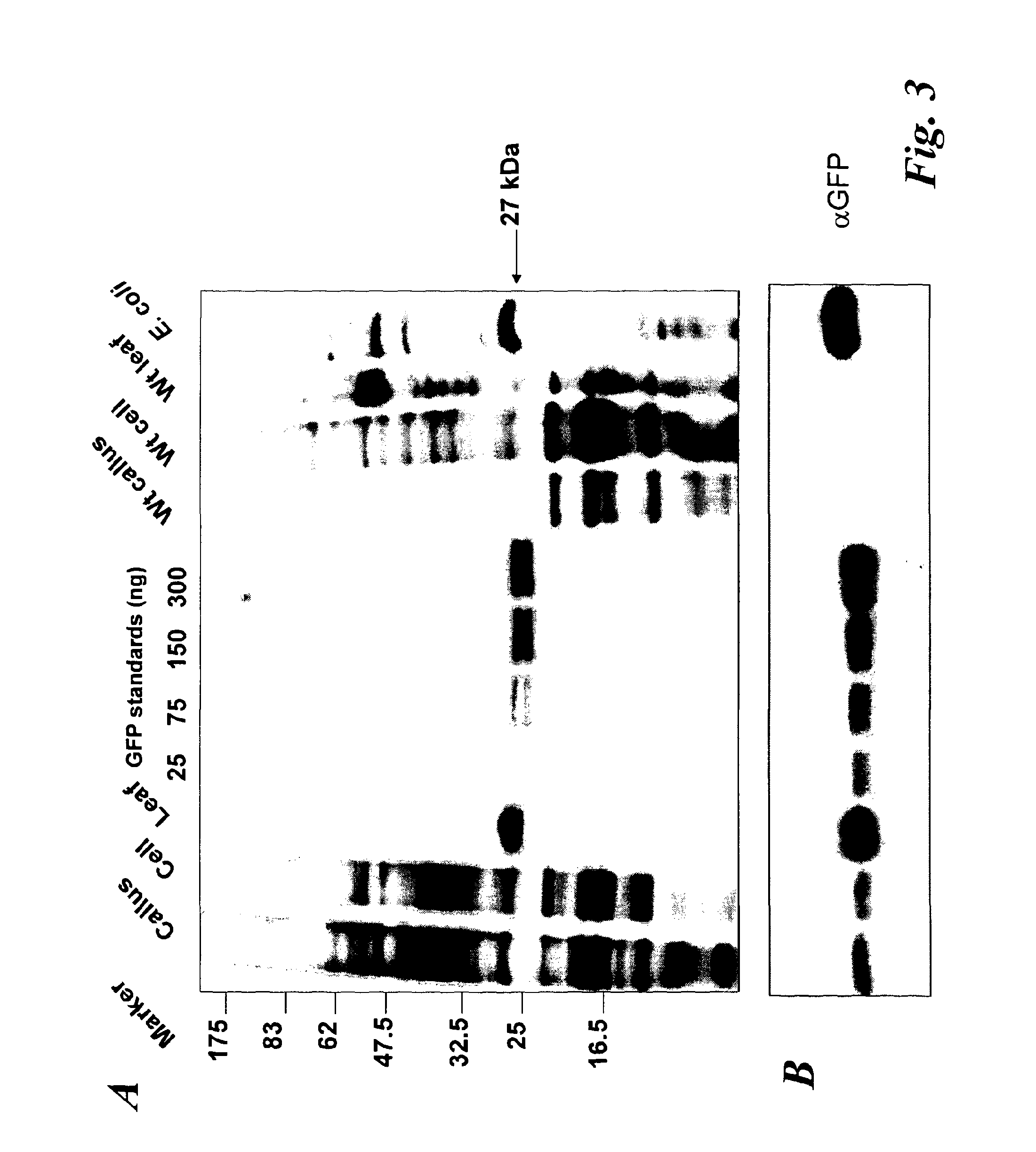
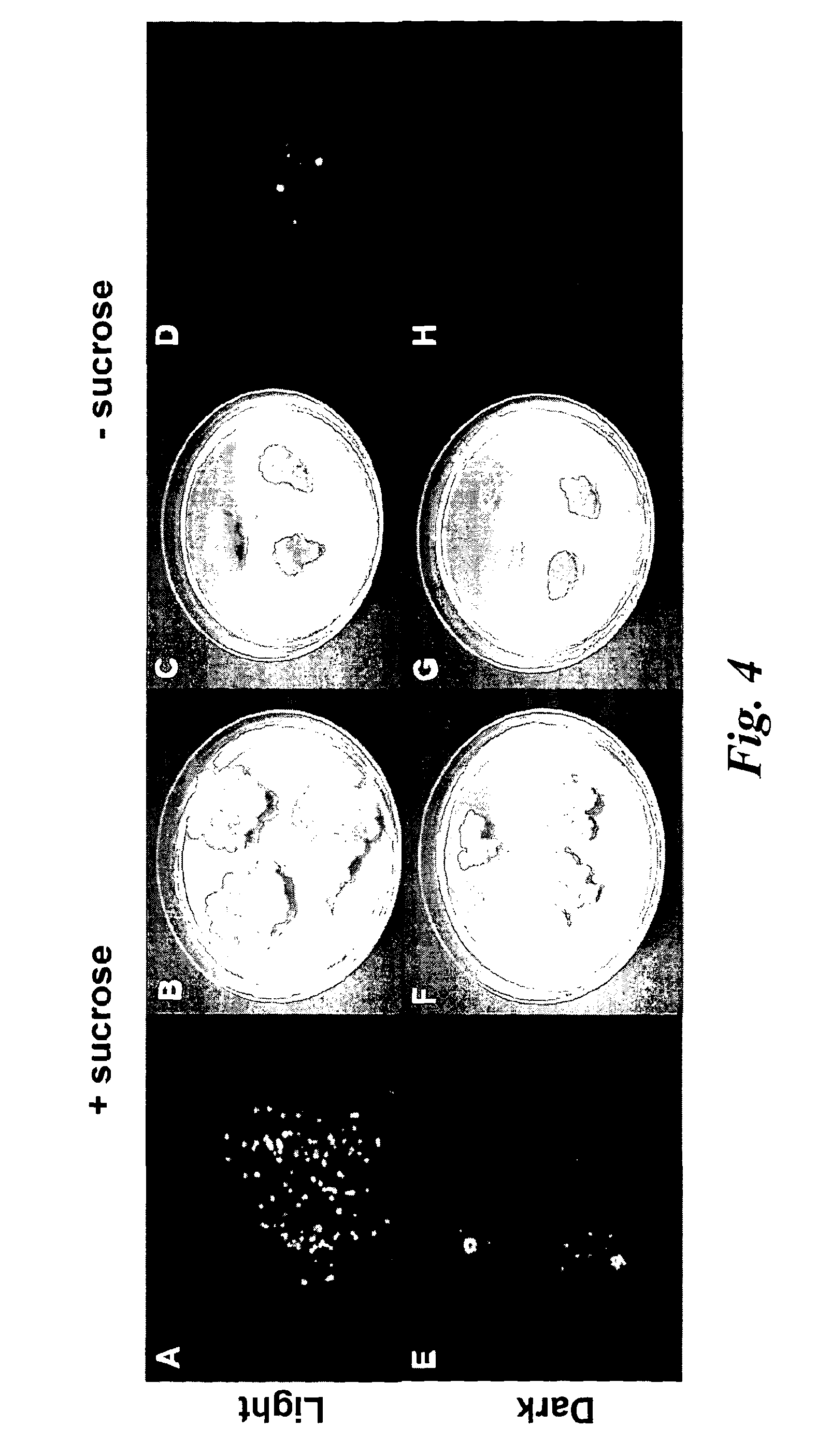
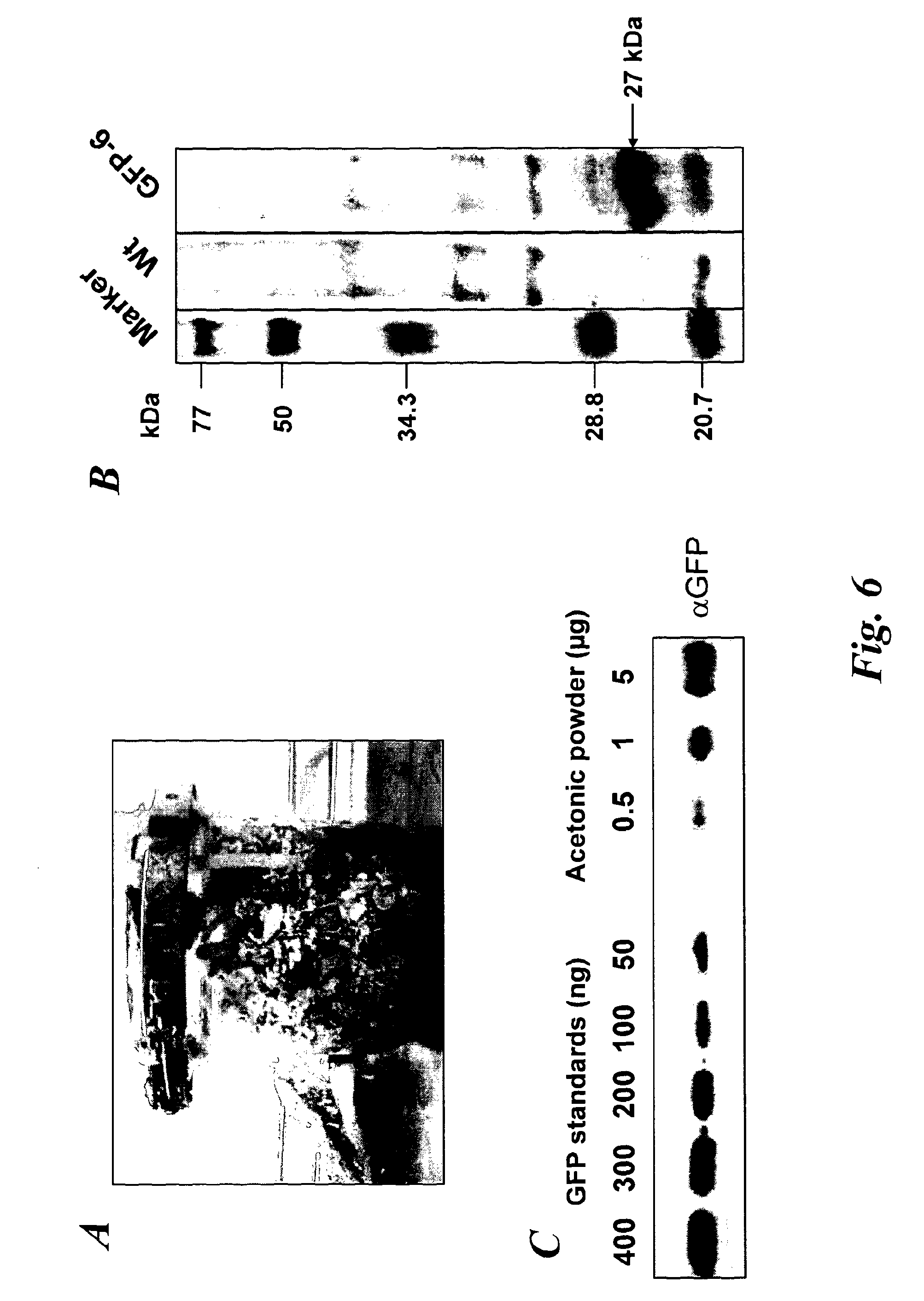
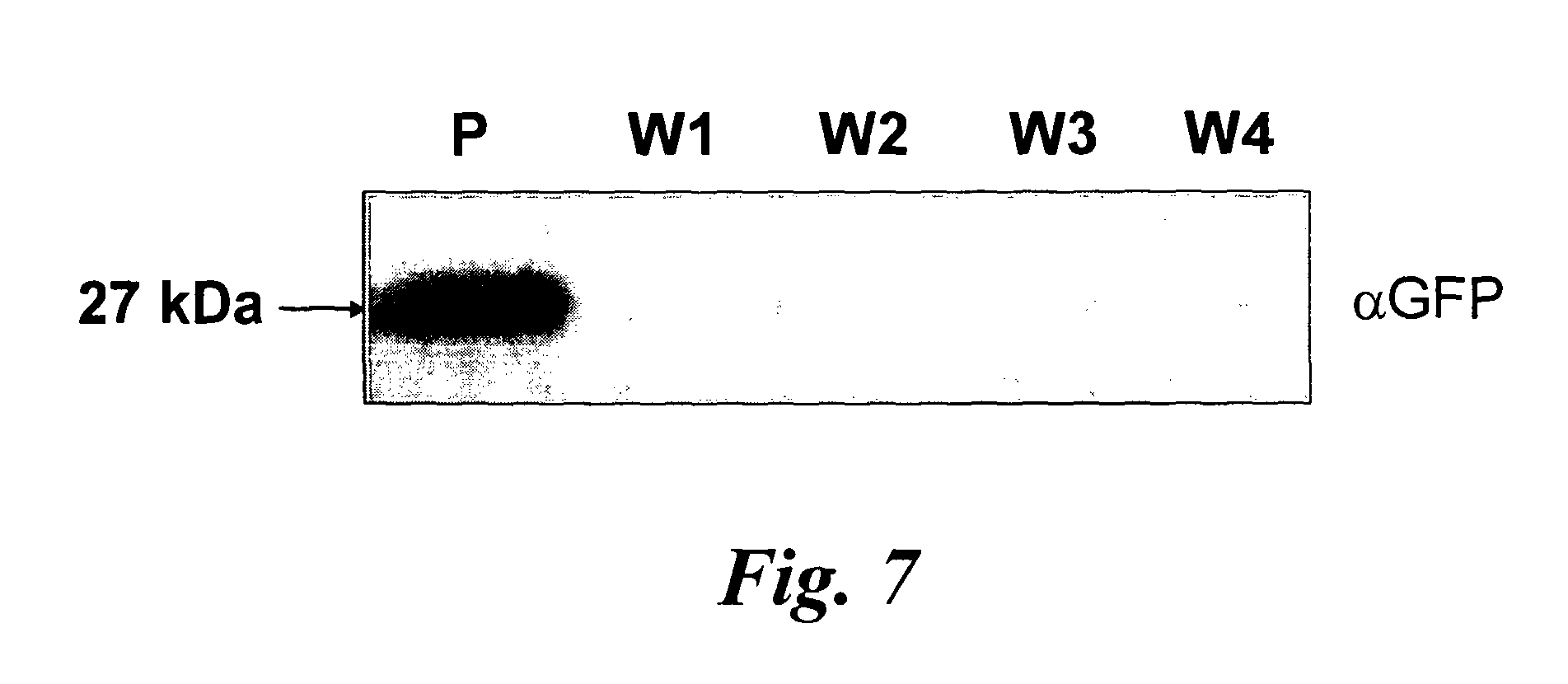
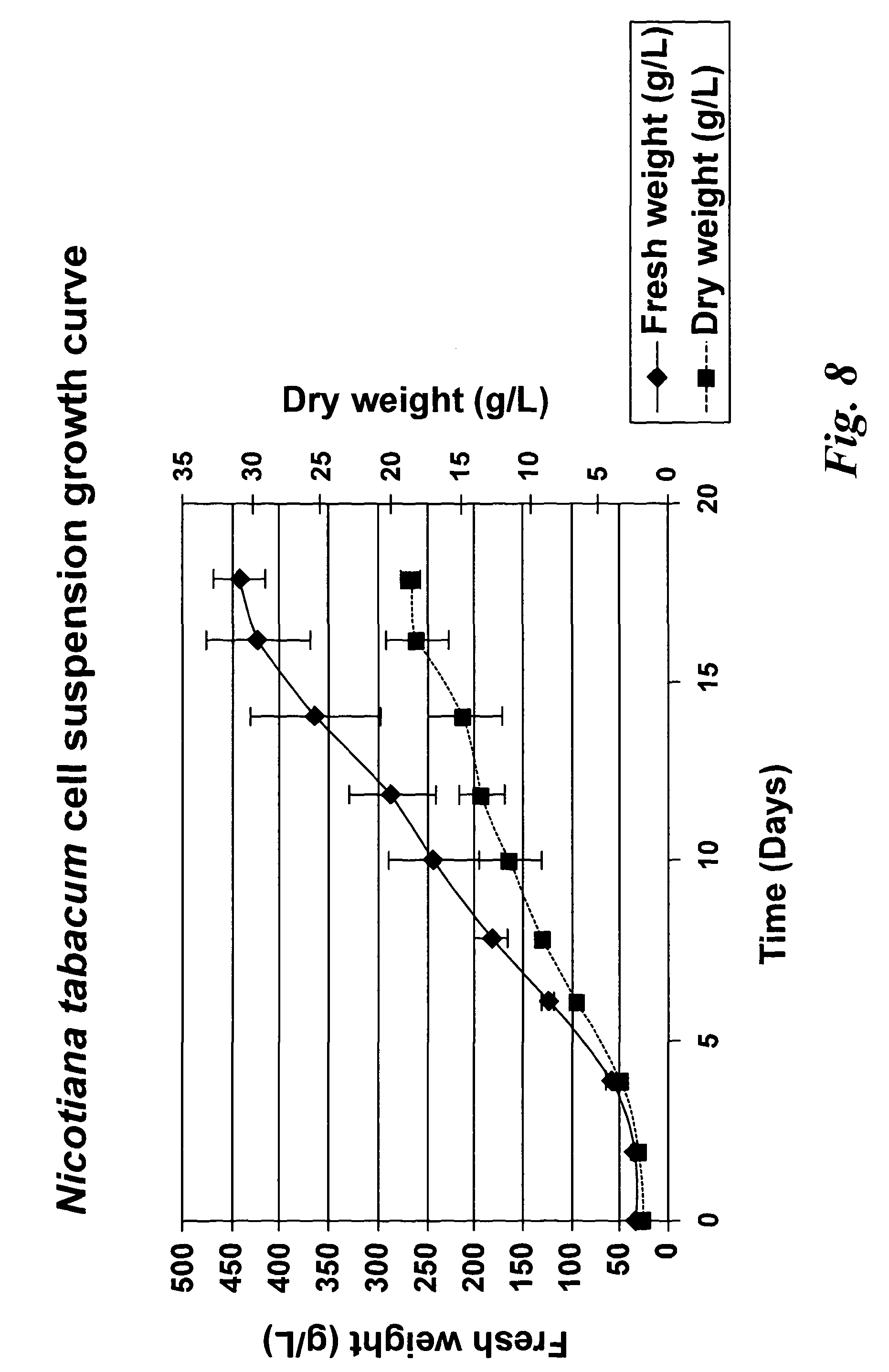
In a further aspect of the invention there is provided a method of capturing carbon dioxide, the method comprising carrying out the method of the first aspect of the invention. A method of purifying a sample comprising exposing the sample to be purified to the leafy biomass derived from the method of the first aspect of the invention. The purification process may be to remove one or more toxins. There is also provided a method of manufacturing a pharmaceutical composition comprising formulating: a component obtained by the methods of the other aspects of the invention and a pharmaceutically acceptable carrier diluent, excipient or carrier. Furthermore, there is provided a pharmaceutical product comprising a component obtained by the methods of the other aspects of the invention and a pharmaceutically acceptable carrier diluent, excipient or carrier. In a further aspect of the invention there is provided a method of manufacturing a biofuel comprising fermentation or transesterification of a component obtained by the methods of the other aspects of the invention. There is also provided a biofuel obtained by this method of manufacture. The present invention will now be described in more detail with reference to the following non limiting Examples and Figures. (A) Physical map of wild-type GFP expression was (A) visualised in the GFP-6 homoplastomic line (GFP-6) under UV and visible light along with control wild-type (wt) tobacco plant. (B) Protein electrophoresis of soluble proteins from GFP-6 and Wt lines. 5 μg of total soluble protein extract of each plant were loaded onto a 12.5% (w/v) SDS-PAGE gel along with prestained protein marker (New England Biolabs, UK) and protein separation was visualised by silver staining. GFP was specifically detected by Western blotting using a specific anti-GFP antibody. Migration of prestained markers is also indicated. Total soluble protein extracts from calli, cell suspensions and leaves from GFP-6 and wild-type tobacco were generated. For calli and cell suspensions, 5 μg total soluble protein were loaded per lane onto a 12.5% (w/v) SDS-PAGE gel whereas only 1 μg was loaded for leaves extracts. (A) corresponds to the silver-stained gel, whereas (B) represents the corresponding Western blot using a GFP antibody. GFP standards were purchased from Roche Life Science, UK and the Prestained Protein Marker from New England Biolabs, UK. The ladder size of the marker proteins are in kDa. Wt stands for Pictures of homoplastomic calli GFP-6 were taken after 4 weeks of growth at 25° C. Plates (A, B, C and D) were grown with 16/8 h light with similar intensity as for tobacco seedlings and (E, F, G and H) were grown in the dark. Only A, B, E and F contained 3% (w/v) sucrose in the media. All media contained 500 mg/L spectinomycin and 500 mg/L streptomycin. Fluorescence emission was detected at 520 nm following excitation at 490 nm using an Axiovert 200 M inverted microscope (Carl Zeiss, Goettingen, Germany) along with the Axiovision software (Version 3.0). Fluorescent exposure was 30 ms, 100 ms and 600 ms for A, D and E, H respectively. Microscope magnification was the same in A, D, E and H at 40×. Total soluble protein were extracted from light (L) or dark (D) grown calli as well as wild-type (Wt) grown under light and sugar. Presence of sucrose in media is indicated by (+) whereas sucrose-free media is described with (−). 5 μg of total soluble protein of the respective calli were loaded onto a 12.5% (w/v) SDS-PAGE gel (L−, L+, D+, D−, wt) and total protein content (A) was detected by silver staining. M represents the Prestained Protein Marker (New England Biolabs, UK) and corresponding sizes are indicated on the left in kDa. (B) GFP+ presence was specifically detected with an anti-GFP antibody. GFP standards (Upstate, USA) were added in the quantities indicated in nanograms. After a 6-weeks incubation period, tobacco biomass of the GFP-6 line (A) was removed from the temporary immersion bioreactor. Total proteins were extracted from newly formed leaves using the acetone extraction protocol and loaded (B) onto a 10% (w/v) SDS-PAGE gel along with prestained SDS-PAGE standard low range (Bio-Rad Laboratories, UK). Proteins from wild-type (wt) and GFP-6 line (GFP-6) were visualised with Coomassie Blue staining. Different dilutions of acetonic powder were analysed by immunoblotting (C) with an anti-GFP antibody and compared to known quantity of GFP protein (Upstate, USA). Western blot representing the GFP presence in several samples from different steps of the acetone extraction protocol. Pellets were resuspended directly in the loading buffer whereas washes were dried overnight in a speedvac (Savant, N.Y., USA) before addition of the loading buffer. Only 5 μl of pellet (P) sample were loaded while all supernatants from washes (W) 1 to 4 were added. Fresh and dry weights of tobacco wild-type cells were determined every 2 days during a 18 day-growth period. Dry weight was measured after leaving fresh tobacco cells 24 h at 80° C. Measurements were done in triplicate. Chloroplast transformation is a promising approach for the commercial production of recombinant proteins in plants. However, gene containment still remains an issue for the large-scale cultivation of transplastomic plants in the field. Here we have evaluated the potential of using tobacco transplastomic cell suspensions for the fully contained production of a model protein, a modified form of the green fluorescent protein (GFP+). In transplastomic leaves GFP+ expression reached approximately 60% of total soluble protein (TSP). Expression in cell suspension cultures (and calli) was much less (1.5% of TSP) but still produced about 7.2 mg per litre of liquid culture. We further investigated the different factors influencing GFP+ production in calli and highlighted the importance of light as an input. Finally we describe the development of a novel protein production platform in which transgenic cell suspension cultures were placed in a temporary immersion bioreactor in the presence of Thidiazuron to initiate shoot formation. GFP+ yield reached an impressive 660 mg per L of bioreactor. This new production platform, combining the rapid generation of transplastomic cell suspension cultures and the use of temporary immersion bioreactors, is a promised route for the fully-contained low-cost production of recombinant proteins. The vector that was constructed to express GFP+ in tobacco chloroplasts is derived from pJST10, which was used to express TetC antigen in tobacco chloroplasts (Tregoning et al, 2003). Plasmid pJST10 targets the insertion of the expression and selection cassette between tobacco chloroplast genes rrn96S and rps12/7 ( To confirm that all chloroplasts of the GFP-6 line were transformed, total genomic DNA was extracted from a leaf of this plant, digested with BglII and subjected to Southern blot analysis ( The tobacco GFP-6 line was grown on soil and expression of GFP+ tested by exposing plants to a UV/blue light source ( The T0 seeds obtained from the GFP-6 line were germinated on MS plates in vitro and the resulting young leaves were used to generate corresponding transplastomic calli and cell suspensions. GFP+ expression was evaluated in the callus state, cell suspension culture and in leaves of the parental plant GFP-6 by SDS-PAGE ( The most striking result of this comparison was the extremely high level of GFP+ expression within tobacco leaves ( In order to assess the importance of light and exogenous sucrose on GFP+ expression, transplastomic calli from the GFP-6 line were grown for one month on Callus Induction Media (CIM) either with or without light and with or without sucrose ( Immunoblotting experiments confirmed that cells grown in complete darkness expressed little or no GFP, whereas in the light, regardless of the presence or absence of sucrose, expression went up ( When grown in the presence of light and sucrose (L+), the level of GFP+ expression was estimated by immunoblotting to be about 4% of TSP ( Given that transplastomic gene expression seemed to be highest in leaf tissue we sought to develop a method for the rapid production of leaf tissue from callus/cell suspensions. In preliminary experiments, we found that addition of Thidiazuron (TDZ), which is known to promote somatic embryo growth in tobacco (Gill and Saxena, 1993), was able to induce shoot formation from GFP-6 calli grown on solid MS medium (data not shown). In order to scale up the production capacity, transplastomic cell suspensions from the tobacco GFP-6 line were loaded into a 2-L bioreactor and temporally submerged in MS media supplemented with 0.1 μM TDZ. After about six weeks, a large number of shoots were produced ( After 40 days, the total biomass was removed from the bioreactor for analysis. Inspection of the plant material revealed the presence of mainly healthy leaves with minimal vitrification. A total amount of about 470 g of fresh weight biomass was produced in the 2-L bioreactor. To evaluate the amount of GFP+ produced within this biomass, a protein precipitation protocol was developed based on protein precipitation in acetone. Using this method, a powder was produced, weighed and loaded onto a SDS-PAGE gel to detect produced GFP+ ( In the bioreactor, total GFP production reached about 660 mg/L at an approximate rate of 17 mg/L/day of GFP over the 40-day growth period. This value is approximately 42-times higher than the rate potentially achievable with cell suspensions of 0.4 mg/L/day. Most work so far in the chloroplast transformation sphere has focussed on leaves for the expression of several genes of interest. Some work has been done on expression in transpiastomic potato tubers (Sidorov et al., 1999) and transplastomic tomato fruit (Ruf et al, 2001) but the expression yields were relatively poor (0.05 and 0.5% of TSP respectively). However, planting transgenic plants, even if they are transplastomic, could still be badly perceived by a large part of the public and the possible environmental issues could have a drastic impact on any future developments. In addition, there are very significant regulatory costs associated with each new transplastomic field releases. Recombinant protein production in contained transplastomic cell based cultures would overcome many of these concerns and should significantly reduce regulatory costs due to the highly contained nature of this new production system. To compare different types of expression system, we first created a homoplastomic line of tobacco that expressed a variant of Green Fluorescent Protein (GFP+). GFP has previously been shown capable of high expression in chloroplasts in a range of different plants including tobacco (Khan and Maliga, 1999; Newell et at, 2003), potato (Sidorov et al, 1999) and lettuce (Kanamoto et al, 2006). The levels of GFP expression described here, approx 60% of TSP in leaves, is at the high end of expression and is similar to the value observed for GFP expression in lettuce where GFP at 36% of TSP was achieved (Kanamoto et al, 2006). Our results showed clearly that levels of GFP+ expression are less in calli and cell suspension cultures compared to leaves ( Expression of GFP+ in transplastomic cell suspensions reached about 1.5% of TSP, which corresponds to 7.2 mg/L at a production rate of 0.4 mg/L/day ( The generally lower expression levels in transplastomic calli and cell suspensions might directly be explained by the choice of the chloroplast transformation vector and specifically by the respective promoter that drove the GFP+ expression. Prrn, the promoter of the RNA16S gene used in pFMGFP, is similar to the RNA16S promoter from rice, whose activity decreased 7 fold in rice embryogenic cells in comparison to its activity in leaves (Silhavy and Maliga, 1998). The same phenomenon might have occurred here since the cell suspension plastids are less differentiated than the leaf chloroplasts. However, further work will have to assess GFP mRNA levels in both leaves and calli to be able to differentiate between a reduction in mRNA levels or a possible variation in chloroplast numbers. Light seemed to be obviously indispensable for significant GFP+ expression ( In our experiments, it was noticeable that the calli and cell suspensions remained green and possessed a large number of chloroplasts widely spread around the cell ( GFP+ production in leaves was vastly superior to that in undifferentiated cells ( However, when transplastomic cell suspensions were placed under temporary immersion conditions where the cell material was subjected to being submerged in liquid occasionally, for only short periods using a temporary immersion type bioreactor, the production of “leafy” material was efficient and significant, with the final biomass production being extremely abundant ( The material was mainly composed of healthy small leaves and the GFP+ content was estimated to reach about 0.66 g/L ( The system described here, properly scaled should be much less labour intensive than the production of whole plants in a green house, and also does not require glasshouse containment facilities. It also offers a potentially faster route to the production of target protein from transformed tissue as seeds do not need to be produced. In fact, once an homoplastomic tobacco line is identified, only one month is required to obtain a cell suspension culture suitable for the temporary immersion bioreactors, whereas, if seeds need to be produced, about 3 months are necessary (Molina et al, 2004). A combination of the temporary immersion growth of transplastomic shoots with recently described disposable bioreactors (Terrier et al, 2007; Ducos et al, 2008) is therefore a promising route for the low-cost production of biopharmaceuticals in plants. Chloroplast transformation vector pFMGFP was created by swapping TeTC gene for gfp+ gene (Scholz et al, 2000) in previously characterized tobacco chloroplast vector pJST10 (Tregoning et al, 2003) by double digestion using NdeI and XbaI restriction sites. Biolistic transformation of 6-weeks old wild-type tobacco leaves with tobacco chloroplast transformation vector pFMGFP was performed on RMOP media (Svab et al, 1990) with a composition based on MS medium supplemented with 1 mg/L thiamine, 100 mg/L myo-inositol, 1 mg/L N6-benzyladenosine (BAP) and 0.1 mg/L 1-Napthaleneacetic acid (NAA) using the PDS1000/He (Bio-Rad, Hercules, Calif., USA) biolistic device with rupture disks of 1100 psi. Vector pFMGFP was coated onto 550 nm gold particles (SeaShell, La Jolla, Calif., USA) according to manufacturer's recommendations. After bombardment, leaves remained in the dark for 48 hours before plant materials were cut into small pieces (5 mm×5 mm) and placed onto RMOP media supplemented with 500 mg/L spectinomycin dihydrochloride. Spectinomycin resistant shoots were subcultured on the same media 4 times. Vector integration into tobacco plastome was evaluated by PCR unsing a primer annealing at the start of gfp+ and the other on the tobacco plastome outside the homologous regions of the vector pFMGFP (data not shown) and transplastomic GFP-6 line was chosen for all further experiments. Homoplastomy state was evaluated by Southern hybridisation of digested total genomic DNA from both wild-type and transplastomic GFP-6 lines. About 7 μg of genomic DNA was digested with BglII and was run on a 0.7% (w/v) agarose gel. The DNA gel was transferred by capillarity onto a nylon membrane (Hybond-N, Amersham, Uppsala, Sweden) overnight in 20×SSC buffer. The probe was DIG-labelled overnight at 37° C. using DIG High Prime DNA Labelling and Detection Starter Kit II (Roche Applied Science, UK). A 3 kb probe homologous to the targeted region was obtained by PCR using primers pJST10-F 5′ AATTCACCGCCGTATGGCTGACCGGCGA 3′ and Rps12-OUT-R 5′ TTCATGTTCCAATTGAACACTGTCCATT 3′ and tobacco genomic DNA as template. Probe labelling and hybridisation were performed according to manufacturer's recommendations with a final probe concentration of 25 ng/ml. Specific signal detection with provided CSPD was detected by X-ray film (Amersham, Uppsala, Sweden) according to the manufacturer's guidelines. After homoplastomy confirmation by Southern blot analysis, GFP-6 plantlet was transferred to soil and allowed to produce seeds. This T0seeds were germinated onto MS media supplemented with 500 mg/L spectinomycin and young T0leaves were used for calli and cell suspensions generation. First, total soluble protein extraction was performed according to (Kanamoto et al., 2006). Plant materials (leaves, calli, cell suspensions) were grounded into a fine powder with liquid nitrogen and mixed with total soluble extraction buffer (50 mM HEPES pH 7.6, 1 mM DTT, 1 mM EDTA, 2% (w/v) polyvinyl pyrrolidone and one tablet of complete protease inhibitors EDTA-free cocktail (Roche Products Ltd, Welwyn Garden City, UK). Plant mixtures were vortexed during 1 min and spun down at 13,000 rpm for 30 min at 4° C. Supernatants were aliquoted and stored at −20° C. until further use. The second method was based on a total protein extraction protocol based acetone precipitation. Plant material was grounded to a fine powder in liquid nitrogen. 30 ml of extraction buffer (80% (v/v) acetone, 5 mM ascorbate) were added to 2 g of plant powder or leaf equivalent and the mixture was homogenised with an Ultra-Turrax (IKA, Heidelberg, Germany) for 15 s on ice. Proteins were pelleted by a centrifugation at 5,000 g for 5 min at 4° C. The supernatant was discarded and the pellet was washed 4 times using the same extraction buffer and same centrifugation conditions. Then the pellet was resuspended in pure acetone and homogenised again. Proteins were spun down once more at 10,000 g for 5 min at 4° C. The supernatant was discarded and the pellet was washed 3 more times in pure acetone. During the last wash, the buffer was aliquoted and dried using a Speed-Vac (Savant, Holbrook, N.Y., USA) and the residual powder was termed acetonic powder. The presence of GFP in the pellet and the different washes was detected by Western blot analysis (Supplementary Proteins from transpiastomic and wild-type samples were resolved in 12.5% (w/v) SDS-PAGE gels along with protein markers and commercially available recombinant GFP (Upstate, Waltham, Mass., USA) for quantification purposes. Protein gels were directly stained with Coomassie Blue or with silver staining. Following electrophoresis, proteins were transferred onto a 0.2 μm nitrocellulose membrane (Bio-Rad, Hercules, Calif., USA) either using the mini Trans-Blot® system (Bio-Rad, Hercules, Calif., USA) or by using the iBlot dry transfer system according to manufacturer's recommendation (Invitrogen, UK). After the transfer, GFP specific detection was performed with primary rabbit polyclonal anti-GFP antibody (provided by Prof Nixon, Imperial College London, UK) diluted 1:20,000 whereas the secondary antibody (Horseradish Peroxidase-conjugated goat anti-rabbit immunoglobulin G, Amersham, Uppsala, Sweden) was diluted 1:10,000. Biochemical detection was performed with the ECL SuperSignal® West Pico Chemiluminescence Substrate kit (Pierce Biotechnology Inc., UK). Tobacco biomass was generated by placing about 7 grams of Transplastomic tobacco calli and cell suspensions expressing GFP and originating from the GFP-6 line were observed using an Axiovert 200 M inverted microscope (Carl Zeiss, Goettingen, Germany) and the Axiovision software (version 3.0). Excitation and emission wavelength were set up at 491 nm and 512 nm respectively, optimal for GFP+ detection (Scholz et al, 2000). Exposures and magnifications varied depending on the experiment and are indicated in each figure. A method for producing leafy biomass from undifferentiated plant cells, the method comprising providing undifferentiated plant cells, contacting them with an agent that promotes differentiation of the cells into leafy tissue and growing the cells in a temporary liquid immersion culture system. This method of the invention may be used to produce polypeptides, and natural medicinal products, and can be used to capture carbon dioxide. A method of producing a polypeptide in plant cells in vitro comprising: providing undifferentiated plant cells containing chloroplasts that carry a transgenic nucleic acid molecule encoding the polypeptide, wherein the plant cells display homoplastomy; and propagating the cells according to the above method to produce leafy biomass containing the polypeptide. 1. A method for producing leafy biomass from undifferentiated plant cells, the method comprising providing undifferentiated plant cells, contacting them with an agent that promotes differentiation of the cells into leafy tissue and growing the cells in a temporary liquid immersion culture system. 2. A method according to 3. (canceled) 4. (canceled) 5. A method according to 6. (canceled) 7. A method according to 8. (canceled) 9. A method according to 10. A method according to 11. A method according to 12. (canceled) 13. A method according to 14. A method according to 15. (canceled) 16. (canceled) 17. A method according to 18. (canceled) 19. A method according to 20. A method according to 21. A method of producing a polypeptide in plant cells in vitro comprising:
providing undifferentiated plant cells containing chloroplasts that carry a transgenic nucleic acid molecule encoding the polypeptide, wherein the plant cells display homoplastomy propagating the cells according to the method of obtaining the polypeptide from the leafy biomass, the polypeptide being a therapeutic polypeptide, an enzyme, a growth factor, an immunoglobulin, a hormone, a structural protein, a protein involved in stress responses of a plant, a biopharmaceutical, or a vaccine antigen. 22. A method according to introducing the transgenic nucleic acid molecule into a chloroplast of a plant cell, inducing the plant cell containing the transgenic nucleic acid molecule to form a callus of undifferentiated cells, and propagating the callus under conditions effective to achieve homoplastomy. 23. A method according to 24. (canceled) 25. (canceled) 26. (canceled) 27. A polypeptide obtained by using the method according to 28. Leafy biomass obtained by the method of 29. A method for obtaining a component present in leafy biomass, the method comprising
producing leafy biomass according to the method of obtaining the component from the leafy biomass, wherein the component is obtained by its secretion by the leafy biomass or by extraction from the leafy biomass. 30. (canceled) 31. A method according to 32. A method according to 33. A system for producing a polypeptide in plant cells in vitro comprising:
an agent which promotes differentiation of undifferentiated cells into leafy tissue; and a nucleic acid molecule encoding the polypeptide, which is adapted for introduction into and expression in chloroplasts. 34. A method of capturing carbon dioxide, the method comprising carrying out the method of 35. A method of purifying a sample wherein one or more toxins are removed comprising exposing the sample to be purified to the leafy biomass derived from the method of 36. (canceled) 37. A method of manufacturing a pharmaceutical composition comprising formulating
a component obtained by the method of 38. A pharmaceutical composition obtained by the method of 39. A method of manufacturing a biofuel comprising fermentation or transesterification of a component obtained by the methods of 40. A biofuel obtained by the method of METHOD
nucleic acid molecule encoding the polypeptide, which is adapted for introduction into and expression in chloroplasts.
EXAMPLE 1
Contained and High-Level Production of Recombinant Protein in Plant Chloroplasts Using a Temporary Immersion Bioreactor
Summary
Results
Generation of Homoplastomic Tobacco Shoots Expressing GFP+
GFP+ Expression in the GFP-6 Line
Comparison of Expression Levels in Leaves, Calli and Cell Suspensions of the GFP-6 Tobacco Line
Influence of Light and Sugar on GFP Expression in Calli
Use of Temporary Immersion Bioreactors for the Production of Transplastomic Biomass
Discussion
Tobacco Transplastomic Cell Suspension Cultures
Factors Influencing the Production of GFP+ in Transplastomic Calli
Transplastomic Biomass Production in Temporary Immersion Bioreactors
Experimental Procedures
Tobacco Shoots, Calli and Cell Suspensions Generation
Construction of Chloroplast Transformation Vector
Generation of Transplastomic Tobacco Plants
Southern Blot Analysis
Protein Extractions
Electrophoresis and Western Blot Analysis
Temporary Immersion Bioreactors
Fluorescence Microscopy
Ratios between fresh, dry weights and acetonic powder. Tissue d.w./f.w. (%) Powder/d.w. (%) Leaves 6.6 ± 0.9 28.3 ± 1.1 Cells 4.4 ± 0.3 14.1 ± 1.4 Calli 3.6 ± 0.4 12.4 ± 1.3 These ratios were calculated for the determination of a robust quantification of GFP. Values represented an average of at least 4 repetitions for fresh weight (f.w.), dry weight (d.w.) and acetonic powder (powder). Calli and cell suspensions (Cells) were harvested at the end of their respective growth phases and leaves measurement was performed on young 2-3 weeks old plantlets (with about 4 leaves per plant, similar to the biomass produced in the temporary immersion bioreactor). REFERENCES